Hydrothermal–thermal conversion synthesis of hierarchical porous MgO microrods as efficient adsorbents for lead(II) and chromium(VI) removal
Linlin Zhanga,
Wancheng Zhu*a,
Heng Zhanga,
Siwei Bia and
Qiang Zhang*b
aDepartment of Chemical Engineering, Qufu Normal University, Shandong 273165, China. E-mail: zhuwancheng@tsinghua.org.cn; Tel: +86-537-4453130
bBeijing Key Laboratory of Green Chemical Reaction Engineering and Technology, Department of Chemical Engineering, Tsinghua University, Beijing 100084, China. E-mail: zhang-qiang@mails.tsinghua.edu.cn
First published on 26th June 2014
Abstract
High crystallinity magnesium oxide (MgO) microrods with hierarchical porous structure (ranging from micropores to mesopores and macropores) and well-defined rod-like morphology have been successfully synthesized via a flux NaCl and surfactant nonyl phenol polyoxyethylene ether (NP-9) directed thermal decomposition of the hydrothermally derived magnesium oxalate dihydrate (MgC2O4·2H2O) microrods. The successive and synergistic effect of NaCl and NP-9 assisted the thermal conversion and promoted the final formation of the well-defined porous MgO microrods with a specific surface area of 50.2 m2 g−1 and well preserved rod-like morphology of the MgC2O4·2H2O precursor. The as-obtained porous MgO microrods were employed as efficient adsorbents for removal of heavy metal ions such as Pb(II) and Cr(VI) from aqueous solutions, and the removal efficiency of Pb(II) (original concentration: 50.0 mg L−1) and Cr(VI) (original concentration: 1.0 mg L−1) was up to 99.5% and 55.6%, respectively. Such well-defined MgO microrods with hierarchical porous structure can also serve as promising candidates for catalyst supports and even as a catalyst themselves in addition to their present waste water treatment applications in various fields.
1. Introduction
The porous metal oxides have been extensively investigated recently owing to their great potential applications in catalysis, adsorption and separation, as well as energy conversion and storage1–3 due to their extraordinary characteristics, such as high surface area, high pore volume, well-defined morphology, and structural flexibility.4,5 Particularly, porous metal oxides with three-dimensional (3D) hierarchical architectures, such as MgO,6,7 Al2O3,8 NiO,9 ZnO,10–12 Fe2O3,13,14 Fe3O4,15 and TiO2,16 assembled by one-dimensional (1D) nanostructures have attracted more and more attention owing to their unique structures and properties.13 The nanometer-sized building blocks interacted with overall micrometer-sized structures exhibit small size effect as well as desirable mechanical strength, easy recovery, and facile transportation.7Among numerous metal oxides, magnesium oxide (MgO) has strongly been considered. As an important inorganic compound, MgO has been widely used as ceramic17 and refractory18 material. In addition, MgO has also been employed as catalyst support for carbon nanotube growth,19,20 catalyst for transesterification,21 and adsorbents for wastewater treatment,22,23 for its distinct advantages such as low cost, and environmental friendliness. Most recently, groundwater contamination by heavy metals and dyes has been a rising worldwide environmental concerns due to its high toxicity to human beings, animals, and environment.24–28 In all efforts toward such concern, MgO has shown excellent adsorption abilities in wastewater treatment for its high surface reactivity and destructive sorbent.29 Venkatesha et al. synthesized MgO nanoparticles (NPs) and employed them for adsorption of reactive and vat dyes, and the maximum adsorption capacities for Levafix Fast Red CA (LFR) and Indanthren Blue BC (IB) were 92.2 and 86.5 mg g−1, respectively.29 Cao et al. obtained flowerlike MgO nanostructures via microwave assisted solvothermal method, which showed superb adsorption capacities for Pb(II) and Cd(II).30
Up to now, many synthetic routes have been developed to synthesize porous MgO structures, such as hydrothermal method,31,32 pulsed-laser deposition,33 in situ chemical vapor deposition5,6 and thermal decomposition of magnesium salts,34,35 or magnesium hydroxide.36–38 Yu et al.36 obtained worm-like porous MgO nanoplates through the calcination of the hydrothermally synthesized Mg(OH)2 precursor. Mesoporous MgO microspheres were acquired by calcination of MgCO3 at 500 °C for 3.0 h, containing three levels (small mesopores, large mesopores and macropores) of hierarchical porous organization.39 Very recently, we have just successfully synthesized hierarchical porous MgO superstructures with a well faceted polyhedron-like profile via a coprecipitation process followed by a facile flux and surfactant directed decomposition.40 However, taking the facile treatments and rational applications into consideration, it is still a challenge to obtain porous MgO nanostructures of high crystallinity with controllable and well-defined 3D hierarchical morphology and porosity.
In this contribution, high crystallinity porous microrods of MgO were synthesized by a facile hydrothermal–thermal conversion process, in which MgO retained the 1D rod-like morphological feature of the precursor during the thermal conversion. Since the thermal decomposition was free of NOx and SOx, magnesium oxalate was selected as the precursor in the present study, which was pre-synthesized via a mild hydrothermal treatment of the MgC2O4·2H2O polyhedrons derived from room temperature coprecipitation.40 During the thermal conversion process, the nonionic surfactant nonyl phenol polyoxyethylene ether (NP-9) and NaCl were employed as the dispersing agent and flux, respectively. The successive and synergistic effect of NP-9 and NaCl favored the final formation of the high crystallinity porous MgO microrods, with a surface area of 50.20 m2 g−1. Additionally, the as-obtained porous MgO microrods were used as high efficiency adsorbents for removal of heavy metal ions such as Pb(II) and Cr(VI) ions from aqueous solutions, indicating the present porous MgO microrods as promising cost effective candidates in the water treatment toward the increasingly focused environmental concerns nowadays.
2. Experimental
2.1. Room-temperature coprecipitation for MgC2O4·2H2O microrods
All reagents were analytical grade and used as received without further purification. The precursor MgC2O4·2H2O microrods were first synthesized via a facile hydrothermal treatment of the MgC2O4·2H2O polyhedrons, which were derived from a room temperature coprecipitation of sodium oxalate (Na2C2O4) and magnesium chloride hexahydrate (MgCl2·6H2O), similar to that reported previously.40 In a typical procedure, keeping the molar ratio of MgCl2 and Na2C2O4 as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 25.0 mmol of Na2C2O4 was dissolved into 25.0 mL of deionized (DI) water, then 12.0 mL of 2.08 mol L−1 MgCl2 solution and extra 3.0 mL of DI water was introduced successively under vigorous magnetic stirring. After stirring for 0.5 h at the room temperature, the resultant white slurry was transferred into a Teflon-lined stainless steel autoclave with a capacity of 67.0 mL. The autoclave was sealed, heated to 180 °C (heating rate: 5 °C min−1), and then kept in an isothermal state for 3.0 h. After cooled down to room temperature naturally, the precursor was obtained after washed with DI water and absolute ethanol for three times and then filtered and finally dried at 70 °C for 6.0 h. In order to investigate the effect of the reaction temperature on the products, the temperature was tuned to room temperature, 60, and 120 °C, respectively, with other conditions unchanged.
:1, 25.0 mmol of Na2C2O4 was dissolved into 25.0 mL of deionized (DI) water, then 12.0 mL of 2.08 mol L−1 MgCl2 solution and extra 3.0 mL of DI water was introduced successively under vigorous magnetic stirring. After stirring for 0.5 h at the room temperature, the resultant white slurry was transferred into a Teflon-lined stainless steel autoclave with a capacity of 67.0 mL. The autoclave was sealed, heated to 180 °C (heating rate: 5 °C min−1), and then kept in an isothermal state for 3.0 h. After cooled down to room temperature naturally, the precursor was obtained after washed with DI water and absolute ethanol for three times and then filtered and finally dried at 70 °C for 6.0 h. In order to investigate the effect of the reaction temperature on the products, the temperature was tuned to room temperature, 60, and 120 °C, respectively, with other conditions unchanged.
2.2. Thermal conversion of MgC2O4·2H2O to form porous MgO microrods
In the subsequent thermal conversion, 1.50 g of the as-obtained precursor MgC2O4·2H2O microrods were ground with 30.0 mmol of NaCl and 8.0 mL of NP-9 for 5.0 and 30.0 min, respectively. The resultant slurry was transferred into a porcelain boat which was subsequently located in a horizontal quartz tube furnace, heated to 650 °C (heating rate: 10 °C min−1) and kept in an isothermal state for 2.0 h. After the thermal conversion, the calcined samples were cooled down to room temperature naturally within the tube furnace, then collected and washed with DI water and absolute ethanol for three times, ultimately filtered and dried at 70 °C for 6.0 h. While to ascertain the specific effect of NaCl and NP-9, the calcination of the precursor without any additives or only in the presence of NaCl was individually carried out at 650 °C within the tube furnace in an air atmosphere, with other conditions kept the same.2.3. Removal of heavy metal Pb(II) and Cr(VI) ions
The as-obtained porous MgO microrods were evaluated for the removal of heavy metal ions of Pb(II) and Cr(VI) from aqueous solutions. In a typical procedure, for each system, 20.0 mg of MgO was dispersed into 20.0 mL of Pb(NO3)2 solution (concentration: 50.0 mg L−1) under vigorous magnetic stirring at room temperature, with the stirring time ranging from 10.0 to 240.0 minutes. After the adsorption systems stirred for their designated time, the resultant slurry was filtered individually, leading to a series of transparent filtrate. The pH value of the filtrate was adjusted to 3.0 using HNO3 (mass concentration: 10%) so as to avoid the hydrolization and thus precipitation of Pb(II), and the filtrate was saved for measurement of the residual Pb2+ within the solution after adsorption by using the porous MgO microrods as adsorbent. In batch experiments for Cr(VI) adsorption, Cr(VI) solution with a concentration of 1.0 mg L−1 was prepared by dissolving specific amount of K2Cr2O7 into proper volume of DI water. All of the Cr(VI) adsorption experiments were conducted by dispersing 20.0 mg of the as-obtained porous MgO microrods into 20.0 mL of the Cr(VI) solution under vigorous magnetic stirring at neutral pH, while with the stirring time ranging from 10.0 to 240.0 min. After the adsorption systems stirred for the designated time, the resultant slurry was filtered individually, resulting in a series of filtrate for further determination of the residual Cr(VI) within the solution after adsorption.In order to investigate the composition change of the adsorbate, the filter cake was collected, washed with DI water and ethanol for three times, and dried at 70 °C for 6.0 h for characterization. For comparison of the adsorption efficiency, analytical grade of MgO reagent was also employed as so-called adsorbent, with other conditions kept the same.
2.4. Characterizations
The phase composition and structure of the samples were identified by an X-ray powder diffractometer (XRD, MiniFlux 600, Rigaku, Japan), using Cu-Kα radiation (λ = 1.54178 Å) and a fixed power source (30.0 kV, 10.0 mA). The morphology and microstructure of the samples were examined by a field emission scanning electron microscopy (SEM, JSM 7401F, JEOL, Japan). The thermal decomposition behavior of the precursor was detected by a thermogravimetric analyzer (TGA, Netzsch STA 409C, Germany), carried out in dynamic air with a heating rate of 10 °C min−1. N2 adsorption–desorption isotherms were measured at 77.3 K using a Chemisorption–Physisorption Analyzer (Autosorb-1-C, Quantachrome, USA). The specific surface area was calculated from the desorption branches in the relative pressure range of 0.05–0.30 using the multipoint Brunauer–Emmett–Teller (BET) method, the pore size distribution was evaluated from the N2 desorption isotherm using the Barrett–Joyner–Halenda (BJH) method, and the total pore volume was calculated for pores with diameters less than a specific value of P/P0 ca. 0.9896. While the actual residual amount of Pb2+ within the solution was determined by an inductively coupled plasma mass spectrometry (ICP-MS) analysis (XSeries 2, Thermo Fisher, USA). The residual concentration of Cr(VI) in the supernatant solution after filtration was measured by diphenylcarbohydrazide method (Chinese GB 7466-87 and GB 7467-87) via a UV-vis spectrophotometer (UV-756CRT, Shanghai Yoke Instruments Meters Co., Ltd., China).3. Results and discussion
3.1. Hydrothermal formation of precursor MgC2O4·2H2O microrods
The chemical constitution, morphology, and decomposition performance of the precursor microrods are showed in Fig. 1. Each diffraction peak of the precursor (Fig. 1(a)) was precisely indexed to that of the standard monoclinic MgC2O4·2H2O (JCPDS no. 26-1223). The narrow and sharp diffraction peaks indicated the high crystallinity of the precursor. This revealed the formation of the precursor MgC2O4·2H2O via the following reaction:| MgCl2 (aq.) + Na2C2O4 (aq.) + 2H2O (l) → MgC2O4·2H2O (s) + 2NaCl (aq.) | (1) |
 | ||
| Fig. 1 (a) XRD pattern, (b) SEM image and (c) TG-DSC curves of the precursor MgC2O4·2H2O microrods hydrothermally synthesized at 180 °C for 3.0 h. | ||
The precursor exhibited rod-like morphology with distinct edges (Fig. 1(b)), which was consistent with the sharp diffraction peaks demonstrated in the XRD pattern. The MgC2O4·2H2O microrods had a length of 5.0–25.0 μm and a width of 1.0–4.0 μm. The thermal behavior of the MgC2O4·2H2O precursor was investigated by the simultaneously recorded thermogravimetric-differential scanning calorimetry (TG-DSC) curves (Fig. 1(c)). The TG curve revealed that the sample mass decreased at a faster rate at 170–250 °C, then abruptly dropped between 400 and 540 °C, and finally remained almost constant thereafter. The former significant mass loss within 170–250 °C was ca. 23.05%, similar to the theoretical value (24.32%) for the dehydration of MgC2O4·2H2O to release two moles of crystal water as shown in eqn (2):
| MgC2O4·2H2O (s) → MgC2O4 (s) + 2H2O (g) | (2) |
The latter abrupt mass loss within 400–540 °C was ca. 46.84%, analogous to the theoretical value (48.65%) for the thermal decomposition of the anhydrous MgC2O4 indicated by eqn (3):
| MgC2O4 (s) → MgO (s) + CO (g) + CO2 (g) | (3) |
Corresponding to the variation of the sample mass with the temperature, the simultaneously recorded DSC profile exhibited two distinct endothermic peaks at 170–250 and 400–540 °C, attributed to the decomposition of the precursor within the two temperature ranges shown as eqn (2) and (3), respectively. The TG-DSC result suggested that the MgO phase could be available when the MgC2O4·2H2O precursor was calcined above 540 °C. The thermal decomposition behavior of the as-synthesized precursor MgC2O4·2H2O microrods was quite similar to that of the previously obtained MgC2O4·2H2O polyhedrons derived from room-temperature coprecipitation.40
Fig. 2 displays the composition and morphology of the precursors synthesized at different temperatures with other parameters kept constant. Notably, with the temperature increasing from room temperature to 120 °C, detectable variation of the crystalline phase has emerged in the corresponding XRD patterns (Fig. 2(a)). The room temperature coprecipitation led to monoclinic glushinskite MgC2O4·2H2O (JCPDS no. 28-0625) submicron polyhedrons (Fig. 2(a1 and b)) with a unit cell = 12.675 × 5.406 × 9.984 Å, and the size was somehow smaller than that of we previously reported MgC2O4·2H2O polyhedrons.40 When the temperature was increased to 60 °C, the product became short rod-like monoclinic MgC2O4·2H2O (JCPDS no. 26-1223) micron particles with a unit cell = 12.689 × 5.391 × 9.977 Å (Fig. 2(a2 and c)). With the increase in the temperature from 60 to 120 °C, the length and aspect ratio of the rod-like micron particles became larger (Fig. 2(a3 and d)). Thus apparently, the hydrothermal treatment brought about the subtle conversion of the crystal structure of the precursor from MgC2O4·2H2O (JCPDS no. 28-0625) submicron particles (Fig. 2(b)) to MgC2O4·2H2O (JCPDS no. 26-1223) short rod-like micron particles (Fig. 2(c)) firstly, and then promoted the elongated or 1D preferential growth, leading to longer (Fig. 2(d)) and even higher aspect ratio (Fig. 1(b)) microrods thereafter.
 | ||
| Fig. 2 (a) XRD patterns and (b–d) SEM images of the precursor MgC2O4·2H2O microrods hydrothermally synthesized at different temperatures for 3.0 h. Temperature (°C): (a1 and b) room temperature; (a2 and c) 60; (a3 and d) 120. | ||
3.2. Thermal conversion of precursor into porous MgO microrods
To improve the crystallinity of the product via the thermal conversion synthesis, the present precursor MgO microrods were calcined at a relatively high temperature as 650 °C. As shown in Fig. 3, after calcined for 2.0 h, all the products were readily indexed as the pure phase of periclase MgO (JCPDS no. 45-0946, Fig. 3(a)). With the introduction of onefold additive NaCl and even two kinds of additives NaCl and NP-9, the crystallinity of the calcined samples became higher and higher. When calcined in the absence of any additive, the products exhibited short rod-like micron and quasi irregular submicron particles (Fig. 3(b)). While calcined in the presence of NaCl, which was used as high-temperature solvent elsewhere,41 the product turned out to be tiny NPs (Fig. 3(c)). The key role of NaCl in improving the crystallinity of the calcined product here was analogous to that found in the flux assisted thermal conversion synthesis of pore-free Mg2B2O5 nanowhiskers.42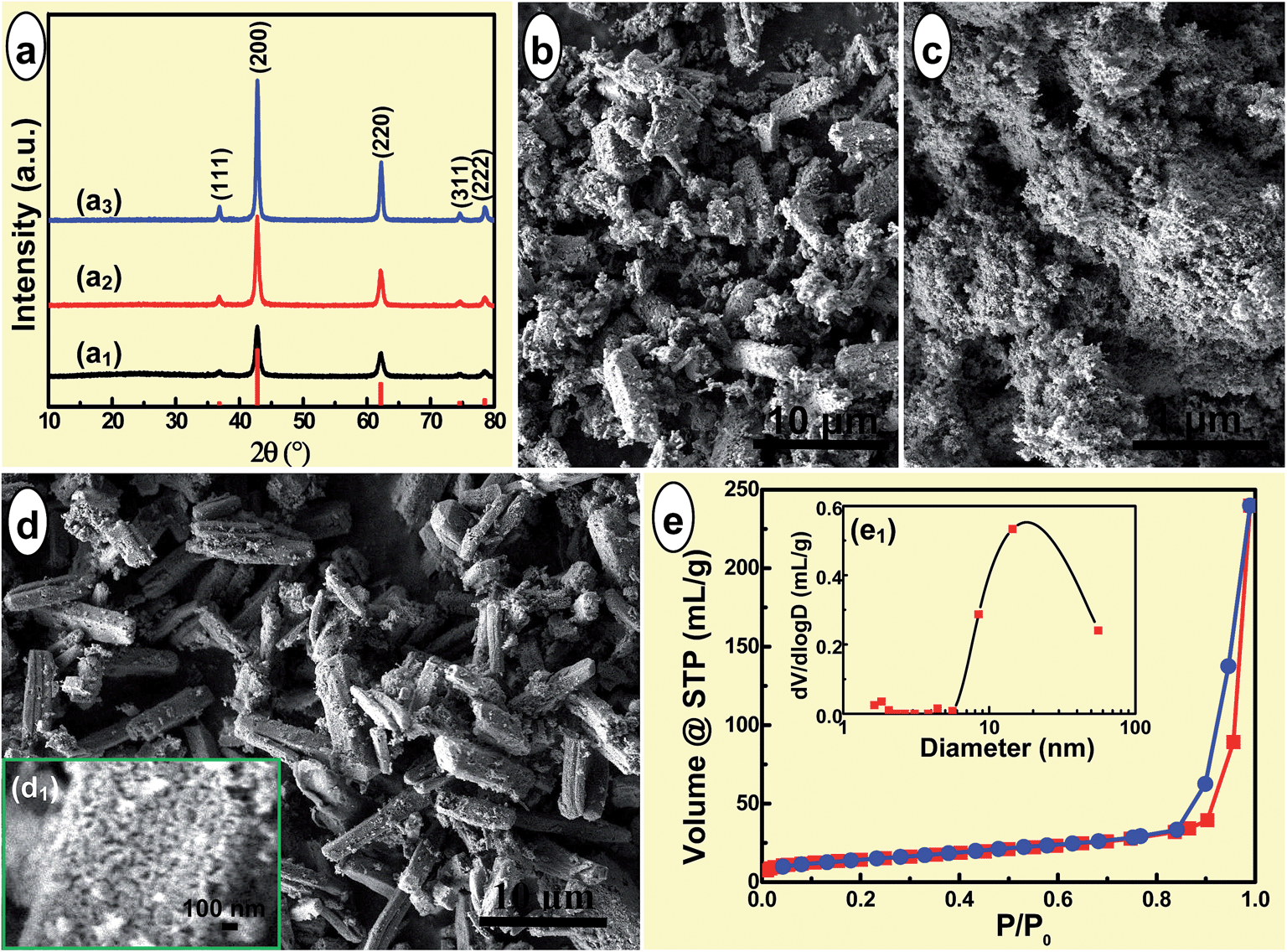 | ||
| Fig. 3 (a) XRD patterns, (b–d) SEM images, and (e) N2 adsorption–desorption isotherms of the products calcined from the precursor in the (a1 and b) absence, or (a2 and c) presence of NaCl, as well as (a3, d, e and e1) NaCl and NP-9 at 650 °C for 2.0 h. Inset (e1) was the corresponding pore size distribution. | ||
Inspired by the striking effect of surfactants and molten salt on the preparation of the 1D metal oxides nanostructures with excellent morphology control,43,44 NaCl and NP-9 were employed in the thermal conversion of the present precursor for MgO as flux and surfactant, respectively. As shown in Fig. 3(d), the product was found as well faceted microrods with some tiny NPs attached on the surfaces. The high magnification SEM image (Fig. 3(d1)) clearly indicated the calcined MgO microrods of porous structure. The calcined porous MgO microrods have well retained the 1D morphology of the precursor MgC2O4·2H2O microrods, and the mutual effect of NaCl and NP-940 on the morphology preservation was similar to that exhibited in formation of the peanut-shaped α-Fe2O3 structures45 as well as rod-like Ni3(BO3)2 particles.46
The porous structure of the MgO microrods was further investigated by N2 adsorption–desorption isotherms. As shown in Fig. 3(e), the N2 adsorption–desorption isotherms presented nearly type III characteristic with a hysteresis loop at a relative high pressure as P/P0 = 0.84–1.0, revealing the existence of the mesoporous47 and macroporous9,48 structure. The present N2 sorption isotherms were similar to those of the porous MgO superstructures,40 and also analagous to those of the mesoporous MgO microspheres39 and porous MgO nanoplates.49 Meanwhile, the pore size distribution (PSD, Fig. 3(e1)) exhibited that the majority of the pores had a pore diameter of 8–50 nm, in addition to a few pores with the pore diameter less than 2 nm or larger than 50 nm. The PSD reconfirmed the as-obtained porous MgO microrods of hierarchical porous structures, ranging from micropores to mesopores and macropores. The multipoint BET was calculated as 50.2 m2 g−1, and this was a slightly smaller than that of MgO microspheres modified with ethylene glycol (55.9 m2 g−1),50 whereas larger than that of the flower-like MgO (45.0 m2 g−1)51 obtained via an ethylene-glycol-mediated self-assembly process, and even larger than the double of that for the porous MgO superstructures.40 This revealed the abundant porosity in the porous MgO microrods.
3.3. Formation mechanism of porous MgO microrods
Based on the above room-temperature coprecipitation especially the hydrothermal conversion and calcination results, the formation mechanism of the present porous MgO microrods was inferred as Fig. 4. At room temperature first, the coprecipitation of Mg2+ and C2O42− led to the nucleation and growth of MgC2O4·2H2O (JCPDS no. 28-0625) submicron polyhedrons (Fig. 4(a)). Then the subsequent hydrothermal treatment promoted the crystalline structure conversion of MgC2O4·2H2O (JCPDS no. 28-0625) polyhedrons, resulting in the hydrothermal formation of the precursor MgC2O4·2H2O (JCPDS no. 26-1223) microrods (Fig. 4(b)) probably due to its intrinsic anisotropic crystal structure. This was similar to the hydrothermal growth of the 1D MgBO2(OH) nanowhiskers without any additive.52 During the calcination, the precursor MgC2O4·2H2O microrods dehydrated and decomposed, and the released H2O, CO as well as CO2 gases led to hierarchical porous structure within the calcined microrods. Undoubtedly, the presence of NaCl and NP-9 was crucial for the preservation of the 1D morphology of the precursor microrods. | ||
| Fig. 4 The proposed formation mechanism of the hierarchical porous MgO microrods. (a) Room-temperature coprecipitation of Mg2+ and C2O42− resulted in the nucleation and growth of MgC2O4·2H2O polyhedrons; (b) hydrothermal treatment of the MgC2O4·2H2O polyhedrons at 180 °C for 3.0 h led to MgC2O4·2H2O microrods; (c) NaCl and NP-9 assisted thermal conversion of the MgC2O4·2H2O microrods at 650 °C for 2.0 h gave rise to porous MgO microrods. | ||
In our previous work, the thermal decomposition phenomena of NP-9 and also synergistic effect of NP-9 and NaCl on the formation of the well faceted hierarchical porous MgO superstructures was investigated in detail.40 Herein very similarly, although calcined at a temperature of 650 °C lower than its normal melting point (ca. 800 °C), NaCl vapor could form44 and then penetrate into and further migrate within the porous structure. The migrating NaCl vapor could serve as the gaseous flux, favoring the in situ crystallization and thus enhancement of the strength of the pore walls within the porous microrods. Consequently, the migrating gaseous flux strengthens the toughness of the backbone and finally favors the preservation of the precursor's original microrod-like morphology. The similar key role of NaCl has also been confirmed in the NaCl assisted formation of the pore-free Mg2B2O5 nanowhiskers via the thermal conversion of MgBO2(OH) nanowhiskers at 650–700 °C.42 And as a nonionic surfactant, NP-9 served as the dispersing agent and thus prevented the porous MgO particles from aggregating with each other (Fig. 4(c)). Consequently, it was the successive and synergistic effect of NaCl and NP-9 that gave rise to the final porous MgO microrods whereas with well faceted profile preserved. Notably, since the standard Gibbs free energy of formation of the MgC2O4·2H2O precursor was not available at present, it was not likely to discuss the above formation mechanism from a pure thermodynamic point of view. However, it was not difficult to understand the thermodynamic feasibility for the whole process, taking the rapid room temperature coprecipitation of MgC2O4·2H2O phase and its thermal decomposition at relatively high temperature to form porous MgO microrods into consideration.
3.4. MgO as adsorbents for removal of Pb(II) and Cr(VI) ions
Since the as-prepared hierarchical porous MgO microrods contained plenty of mesopores and macropores, the corresponding application as absorbents for removal of lead(II) and chromium(VI) heavy metal ions was evaluated. Fig. 5(a) shows the adsorption rate of Pb(II) ion with an initial concentration of 50.0 mg L−1. The adsorption process was very rapid during the first 30 min that the concentration of Pb(II) ion was decreased to 0.21 mg L−1 at 10 min, and the equilibrium was established within 1.0 h. After 4.0 h, the concentration of Pb(II) ion was 0.15 mg L−1. Correspondingly, the ratio of the concentration of Pb(II) at specific adsorption time to that of the original one decreased from 1.0 to 0.007 within 10 min, kept a slight dynamic vibration thereafter and reached equilibrium within 1.0 h. This indicated the adsorption efficiency has reached 99.3% within 10 min, and ultimately remained relatively constant at 99.5%. This was similar to 90–95% of EDTA–graphene oxide (initial concentration: 100 mg L−1),27 whereas much higher than 70% of the adsorbent Ca–montmorillonite (initial concentration: 58.79 mg L−1)53 and 70% of activated carbon–chitosan complex (initial concentration: 0.025 mol L−1).54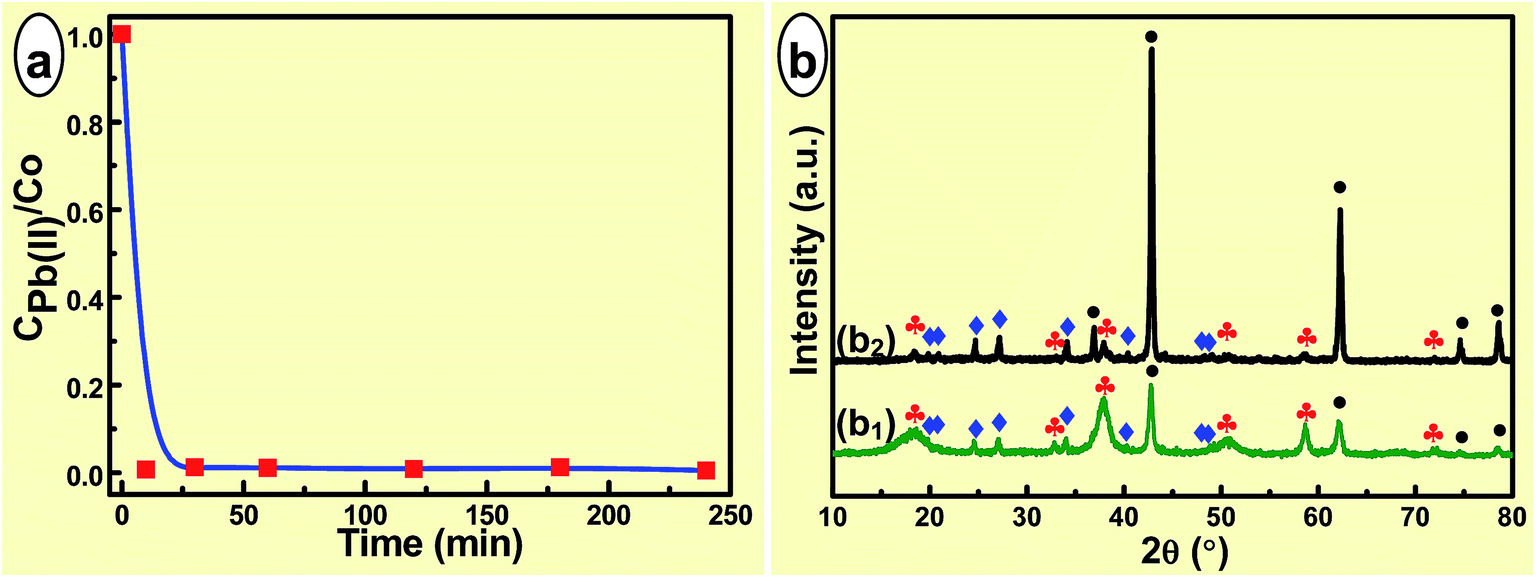 | ||
Fig. 5 (a) The adsorption of Pb(II) and (b) the compositions of adsorbates after adsorption, using (a and b1) the as-synthesized hierarchical porous MgO microrods as adsorbent and (b2) MgO reagent for comparison. Initial concentration (mg L−1): Pb(II)-50.0. ● MgO (JCPDS no. 45-0946),  Mg(OH)2 (JCPDS no. 07-0239), Mg(OH)2 (JCPDS no. 07-0239),  Pb3(CO3)2(OH)2 (JCPDS no. 13-0131). Pb3(CO3)2(OH)2 (JCPDS no. 13-0131). | ||
In order to better understand the present high adsorption efficiency, the XRD patterns of adsorbates were recorded (Fig. 5(b1)). As shown, all the diffraction peaks could be assigned to MgO (JCPDS no. 45-0946), Mg(OH)2 (JCPDS no. 07-0239) and Pb3(CO3)2(OH)2 (JCPDS no. 13-0131). The Mg(OH)2 phase should be attributed to the hydration of MgO in aqueous solution. During the adsorption process, partially dissolved CO2 reacted with MgO and Pb(II), producing Pb3(CO3)2(OH)2. In order to verify the reaction between MgO, CO2 and Pb(II), commercial MgO reagent was also employed as adsorbent to undergo the same adsorption process, which also brought about the Pb3(CO3)2(OH)2 phase (Fig. 5(b2)). Thus, the adsorption process was physical adsorption based on the hierarchical porous nature therein and the chemical adsorption, which was similar to the reaction between the flowerlike MgO and Pb(II) in the course of adsorption.30
To further ascertain the formation of Pb3(CO3)2(OH)2 phase, extended experiments were carried out. However, where did carbonate come from or how was it formed was a hard nut to crack. Even the same adsorption test was performed in a strict N2 protected system, the emergence of Pb3(CO3)2(OH)2 was also confirmed. Taking the basic groups on the surfaces of MgO and relative solubility of CO2 in solution into account, the carbonate was probably derived from the adsorbed acidic CO2 into the basic hierarchical porous MgO microrods from air and that dissolved into the solution. As known, the solubility product constant Ksp values of magnesium carbonate and lead carbonate were 3.5 × 10−8 and 7.4 × 10−14, respectively. Although the Ksp of Pb3(CO3)2(OH)2 was not available at present, it was believed smaller than that of lead carbonate according to the experimental results, and also much smaller than that of magnesium carbonate. Thus, during the adsorption process the adsorbed CO2 within the porous MgO microrods and dissolved CO2 within the solution might be released and reacted with Pb(II) under vigorous magnetic stirring, resulting in the Pb3(CO3)2(OH)2 phase.
The as-obtained hierarchical porous MgO microrods were also employed as the adsorbents for removal of Cr(VI) (initial concentration: 1.0 mg L−1), as shown in Fig. 6(a), with MgO reagent for comparison (Fig. 6(b)). Obviously, the as-obtained porous MgO microrods displayed higher adsorption efficiency than MgO reagent. About 30% and 10% of Cr(VI) was removed by 1.0 h of adsorption by using the as-obtained porous MgO microrods and MgO reagent as adsorbents, respectively. While after 4.0 h, the adsorption efficiencies was increased to 55.6% and 20.7%, respectively, indicating the significant effect of the hierarchical porous structures within the as-obtained MgO microrods on the adsorption of Cr(VI). The adsorption efficiency of Cr(VI) on the present hierarchical porous MgO microrods was lower than that on the maghemite (Fe3O4) NPs (58.9%, pH = 2.5, initial concentration: 150 mg L−1),55 whereas similar to that on the active carbon supported MgO micron particles (11.14%, pH = 2, initial concentration: 50 mg L−1),56 taking the difference of initial concentration employed into consideration. Notably however, the adsorption efficiency of Cr(VI) was not so remarkable compared with Pb(II). Under the aforesaid neutral pH condition, the degree of protonation of the porous MgO microrods' surface reduced gradually,57 while the dominant ionic form of Cr(VI) is dichromate ion (Cr2O72− and other forms), and the abundance of OH− ions increased the hindrance to diffusion of dichromate ions.37 After adsorption of Cr(VI), the adsorbates were confirmed as MgO (JCPDS no. 45-0946) and Mg(OH)2 (JCPDS no. 07-0239), and no Cr(VI) containing compounds were detected. This explained the relatively low adsorption efficiency of Cr(VI) by the present hierarchical porous MgO microrods, compared with the adsorption of Pb(II).
 | ||
Fig. 6 (a) The adsorption of Cr(VI) and (b) compositions of the adsorbates after adsorption, using (a1 and b1) the as-synthesized hierarchical porous MgO microrods as adsorbents and (a2 and b2) MgO reagent for comparison. Initial concentration (mg L−1): Cr(VI)-1.0. ● MgO (JCPDS no. 45-0946),  Mg(OH)2 (JCPDS no. 07-0239). Mg(OH)2 (JCPDS no. 07-0239). | ||
Although previous literatures showed that acid conditions were favorable for the removal of Cr(VI),55,56 the neutral pH was employed when using pure porous MgO microrods as adsorbents. On one hand, distinct base condition will promote the transformation from MgO to Mg(OH)2, which will definitely destroy the structure and further influence the recycling of the porous MgO microrods as adsorbents. On the other hand, distinct acid condition will favor the dissolution and further loss of MgO as adsorbents. Moreover, acid or base condition will exacerbate the impact to the environment. As a matter of fact, when the adsorption of Cr(VI) was performed within a different pH range of 2.0–8.0, the adsorption rate increased from 12% (pH = 2.2) to 32% (pH = 3.9) and then monotonously decreased to 2% (pH = 8.0). Based on the adsorption performance at such a pH range and especially the impact to the environment, a neutral pH condition for the removal of Cr(VI) should be recommended. After all, the adsorption efficiency can be improved to some extent by using more inexpensive porous MgO microrods as adsorbents.
4. Conclusions
Hierarchical porous MgO microrods with a high crystallinity, well-defined rod-like morphology were obtained via a hydrothermal process (180 °C, 3.0 h) followed by a mild flux NaCl and surfactant NP-9 directed thermal conversion of the precursor at 650 °C. During the hydrothermal treatment, with the increase in the temperature, the room-temperature precipitate MgC2O4·2H2O (JCPDS no. 28-0625) submicron polyhedrons were converted to MgC2O4·2H2O (JCPDS no. 26-1223) firstly, and then the elongated or 1D preferential growth was promoted, leading to higher aspect ratio microrods. Within the subsequent thermal conversion, the successive and synergistic effect of NaCl and NP-9 played the key role in the final formation of the well-defined MgO microrods with a hierarchical porous structure (ranging from micropores to mesopores and macropores), specific surface area of 50.2 m2 g−1 and well preserved rod-like morphology of the precursor MgC2O4·2H2O microrods. The as-obtained hierarchical porous MgO microrods were utilized as efficient adsorbents for removal of heavy metal ions including Pb(II) and Cr(VI) from aqueous solutions, and the removal efficiency of Pb(II) (original concentration: 50.0 mg L−1) and Cr(VI) (original concentration: 1.0 mg L−1) was up to 99.5% and 55.6%, respectively. In addition to the present waste water treatment, the as-synthesized high crystallinity MgO microrods with hierarchical porous structure can also serve as alternative candidates for catalyst support and even catalyst itself in varieties of fields.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 21276141), the State Key Laboratory of Chemical Engineering, China (no. SKL-ChE-12A05). The authors also thank the reviewers for the constructive comments and suggestions on the improvement of the work.References
- A. Vantomme, A. Léonard, Z.-Y. Yuan and B.-L. Su, Colloids Surf., A, 2007, 300, 70–78 CrossRef CAS PubMed.
- K. Assaker, C. Carteret, P. Durand, L. Aranda, M. J. Stébé and J. L. Blin, J. Phys. Chem. C, 2013, 117, 16500–16508 CAS.
- M. Q. Zhao, Q. Zhang, J. Q. Huang and F. Wei, Adv. Funct. Mater., 2012, 22, 675–694 CrossRef CAS.
- L. H. Ai, H. T. Yue and J. Jiang, Nanoscale, 2012, 4, 5401–5408 RSC.
- L.-R. Qin, J.-W. Zhao, L.-D. Zhang and X.-W. Zou, Chem. Lett., 2006, 35, 380–381 CrossRef CAS.
- Y. Yan, L. Zhou and Y. Zhang, J. Phys. Chem. C, 2008, 112, 19831–19835 CAS.
- X.-Y. Yu, T. Luo, Y. Jia, Y.-X. Zhang, J.-H. Liu and X.-J. Huang, J. Phys. Chem. C, 2011, 115, 22242–22250 CAS.
- T.-Z. Ren, Z.-Y. Yuan and B.-L. Su, Langmuir, 2004, 20, 1531–1534 CrossRef CAS.
- P. Tian, J. Ye, G. Ning, W. Gong, N. Xu, Q. Zhang and Y. Lin, RSC Adv., 2012, 2, 10217–10221 RSC.
- H. Bai, Z. Liu and D. D. Sun, Chem.–Asian J., 2012, 7, 1772–1780 CrossRef CAS PubMed.
- M. Abd-Ellah, N. Moghimi, L. Zhang, N. F. Heinig, L. Zhao, J. P. Thomas and K. T. Leung, J. Phys. Chem. C, 2013, 117, 6794–6799 CAS.
- L. F. Xu, Q. W. Chen and D. S. Xu, J. Phys. Chem. C, 2007, 111, 11560–11565 CAS.
- W. Zhu, X. Cui, X. Liu, L. Zhang, J.-Q. Huang, X. Piao and Q. Zhang, Nanoscale Res. Lett., 2013, 8, 2 CrossRef PubMed.
- W. Zhu, X. Cui, L. Wang, T. Liu and Q. Zhang, Mater. Lett., 2011, 65, 1003–1006 CrossRef CAS PubMed.
- J. Liu, J. Cheng, R. Che, J. Xu, M. Liu and Z. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 2503–2509 CAS.
- Y. Li, X. Yan, W. Yan, X. Lai, N. Li, Y. Chi, Y. Wei and X. Li, Chem. Eng. J., 2013, 232, 356–363 CrossRef CAS PubMed.
- T. Kathrina and R. D. Rawlings, J. Mater. Sci., 1997, 32, 3951–3959 CrossRef CAS.
- M.-A. Faghihi-Sani and A. Yamaguchi, Ceram. Int., 2002, 28, 835–839 CrossRef CAS.
- Q. Zhang, M. Zhao, J. Huang, W. Qian and F. Wei, Chin. J. Catal., 2008, 29, 1138–1144 CrossRef CAS.
- Q. Zhang, J.-Q. Huang, W.-Z. Qian, Y.-Y. Zhang and F. Wei, Small, 2013, 9, 1237–1265 CrossRef CAS PubMed.
- G. Zhao, J. Shi, G. Liu, Y. Liu, Z. Wang, W. Zhang and M. Jia, J. Mol. Catal. A: Chem., 2010, 327, 32–37 CrossRef CAS PubMed.
- G. Moussavi and M. Mahmoudi, Chem. Eng. J., 2009, 152, 1–7 CrossRef CAS PubMed.
- C. Gao, W. Zhang, H. Li, L. Lang and Z. Xu, Cryst. Growth Des., 2008, 8, 3785–3790 CAS.
- X. Sun, L. Yang, Q. Li, J. Zhao, X. Li, X. Wang and H. Liu, Chem. Eng. J., 2014, 241, 175–183 CrossRef CAS PubMed.
- M. Eloussaief, W. Hamza, N. Kallel and M. Benzina, Environ. Prog. Sustainable Energy, 2013, 32, 229–238 CrossRef CAS.
- A. K. Bhattacharya, T. K. Naiya, S. N. Mandal and S. K. Das, Chem. Eng. J., 2008, 137, 529–541 CAS.
- C. J. Madadrang, H. Y. Kim, G. Gao, N. Wang, J. Zhu, H. Feng, M. Gorring, M. L. Kasner and S. Hou, ACS Appl. Mater. Interfaces, 2012, 4, 1186–1193 CAS.
- J. Hu, Z. Song, L. Chen, H. Yang, J. Li and R. Richards, J. Chem. Eng. Data, 2010, 55, 3742–3748 CrossRef CAS.
- T. G. Venkatesha, R. Viswanatha, Y. Arthoba Nayaka and B. K. Chethana, Chem. Eng. J., 2012, 198–199, 1–10 CrossRef CAS PubMed.
- C.-Y. Cao, J. Qu, F. Wei, H. Liu and W.-G. Song, ACS Appl. Mater. Interfaces, 2012, 4, 4283–4287 CAS.
- Z. Zhao, H. Dai, Y. Du, J. Deng, L. Zhang and F. Shi, Mater. Chem. Phys., 2011, 128, 348–356 CrossRef CAS PubMed.
- Y. Liu, Q. Li, S. Gao and J. K. Shang, J. Am. Ceram. Soc., 2011, 94, 217–223 CrossRef CAS PubMed.
- B. Huo, L. Hu, H. Zhang, Q. Fu, J. Zhu and X. Chen, Micro Nano Lett., 2009, 4, 59–62 CAS.
- M. Sharma and P. Jeevanandam, J. Alloys Compd., 2011, 509, 7881–7885 CrossRef CAS PubMed.
- N. Sutradhar, A. Sinhamahapatra, S. K. Pahari, P. Pal, H. C. Bajaj, I. Mukhopadhyay and A. B. Panda, J. Phys. Chem. C, 2011, 115, 12308–12316 CAS.
- J. C. Yu, A. Xu, L. Zhang, R. Song and L. Wu, J. Phys. Chem. B, 2004, 108, 64–70 CrossRef CAS.
- R. Hahn, J. G. Brunner, J. Kunze, P. Schmuki and S. Virtanen, Electrochem. Commun., 2008, 10, 288–292 CrossRef CAS PubMed.
- Y. Ding, G. Zhang, H. Wu, B. Hai, L. Wang and Y. Qian, Chem. Mater., 2001, 13, 435–440 CrossRef CAS.
- J. Zhou, S. Yang and J. Yu, Colloids Surf., A, 2011, 379, 102–108 CrossRef CAS PubMed.
- W. Zhu, L. Zhang, G.-L. Tian, R. Wang, H. Zhang, X. Piao and Q. Zhang, CrystEngComm, 2014, 16, 308–318 RSC.
- A. L. Tiano, A. C. Santulli, C. Koenigsmann, M. Feygenson, M. C. Aronson, R. Harrington, J. B. Parise and S. S. Wong, Chem. Mater., 2011, 23, 3277–3288 CrossRef CAS.
- W. C. Zhu, Q. Zhang, L. Xiang and S. L. Zhu, Cryst. Growth Des., 2011, 11, 709–718 CAS.
- W. Wang and L. Ao, Cryst. Growth Des., 2008, 8, 358–362 CAS.
- P. M. Rørvik, T. Lyngdal, R. Sæterli, A. T. J. van Helvoort, R. Holmestad, T. Grande and M. A. Einarsrud, Inorg. Chem., 2008, 47, 3173–3181 CrossRef PubMed.
- T.-J. Park and S. S. Wong, Chem. Mater., 2006, 18, 5289–5295 CrossRef CAS.
- X. Liu, W. Zhu, X. Cui, T. Liu and Q. Zhang, Powder Technol., 2012, 222, 160–166 CrossRef CAS PubMed.
- J. Lian, C. Zhang, P. Wang and D. H. L. Ng, Chem.–Asian J., 2012, 7, 2650–2655 CrossRef CAS PubMed.
- F. Mou, J. Guan, H. Ma, L. Xu and W. Shi, ACS Appl. Mater. Interfaces, 2012, 4, 3987–3993 CAS.
- H. X. Niu, Q. Yang, K. B. Tang and Y. Xie, Microporous Mesoporous Mater., 2006, 96, 428–433 CrossRef CAS PubMed.
- J. Jin, Z. Zhang, H. Ma, X. Lu, J. Chen, Q. Zhang, H. Zhang and Y. Ni, Mater. Lett., 2009, 63, 1514–1516 CrossRef CAS PubMed.
- S. W. Bain, Z. Ma, Z. M. Cui, L. S. Zhang, F. Niu and W. G. Song, J. Phys. Chem. C, 2008, 112, 11340–11344 CAS.
- W. C. Zhu, S. L. Zhu and L. Xiang, CrystEngComm, 2009, 11, 1910–1919 RSC.
- Y. Li, J. Wang, X. Wang and J. Wang, Ind. Eng. Chem. Res., 2012, 51, 6520–6528 CrossRef CAS.
- H. Ge and X. Fan, Chem. Eng. Technol., 2011, 34, 1745–1752 CrossRef CAS.
- J. Hu, G. Chen and I. M. C. Lo, Water Res., 2005, 39, 4528–4536 CrossRef CAS PubMed.
- K. Peng, Z. Jia, R. Zhu and J. Zhou, Journal of The Chinese Ceramic Society, 2011, 39, 1651–1658 CAS.
- M. Kobya, Bioresour. Technol., 2004, 91, 317–321 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |