DOI:
10.1039/C4RA03754E
(Paper)
RSC Adv., 2014,
4, 29957-29963
Bi2O3 and BiOCl electrospun nanosheets and morphology-dependent photocatalytic properties†
Received
24th April 2014
, Accepted 12th June 2014
First published on 12th June 2014
Abstract
BiOCl and Bi2O3 nanosheet like structures were produced by electrospinning. The morphological changes were observed by changing precursor (BiOCl3 and Bi(NO3)3·5H2O) concentrations. These nanosheets were analyzed by XRD, which reveals that the crystal structures of BiOCl and Bi2O3 belonged to tetragonal and beta-phase systems respectively. Both nanostructures were employed for the photodegradation of Alizarin Red S (ARS) dye under UV light (<390 nm) irradiation. BiOCl nanosheet like structures exhibited superior photocatalytic activity (PCA) for the degradation of ARS dye and their half-life was estimated from the kinetic plots of PCA. A plausible reaction mechanism is proposed for the PCA and discussed in detail.
Introduction
In recent years, novel layered/sheet-like structures of bismuth oxyhalides BiOX (X = Cl, Br, and I) have drawn much attention due to their optical properties and promising industrial applications.1 Bismuth oxychloride (BiOCl) and bismuth oxide (Bi2O3) have potential for degradation of organic dyes2,3 and non-biodegradable synthetic dyes.4,5 BiOCl exhibits the tetragonal space group P4/nmm and consists of stacked sheets of Cl–Bi–O–Bi–Cl held together by the nonbonding interaction through the Cl atoms along the c-axis.6 In each sheet, a bismuth center is surrounded by four oxygen (O2) and four chlorine (Cl) atoms in an asymmetric decahedral geometry. These tetragonal crystal structures of BiOCl can be flattened into sheet-like structures with a high aspect ratio. BiOCl had been used in several applications2,7,8 because the strong intralayer bonding and the weak interlayer interactions (van der Waals) between the sheets would be favourable for work on highly anisotropic structures, as well as for improved electrical, optical, and mechanical properties. Owing to the unique sheet-like structure and high photocorrosion stability in the presence of redox couples, BiOCl has recently been used for photocatalysis9,10 and in photoelectrochemical (PEC) applications.2,8,11 Zhang et al.2 reported that BiOCl exhibited better photocatalytic activity (PCA) on the methyl orange (MO) dye degradation, than did TiO2 (P25, Degussa). On the other hand, Bi2O3 was proved to be a significant photocatalyst for the degradation of rhodamine B (RB) and MO dye under UV and visible-light irradiation, respectively.3,5,12 Lin et al.13 reported that the PCA of Bi3O4Cl (synthesized from BiOCl and Bi2O3 via a solid state reaction) was more effective for the degradation of MO dye than that of anatase TiO2 under UV light irradiation.
In addition, low-dimensional nanostructures14–16 could enhance the PCA enormously and, therefore, the preparation of nanostructured BiOCl and Bi2O3 are considered promising for photocatalytic applications. The nanoparticular BiOCl and Bi2O3 can be an efficient photocatalyst in decomposing methyl orange under UV irradiation.2,3 However, bismuth oxide nanoparticles have limitations; for example, suspended nanoparticulate catalysts are easily lost in the process of the PCA reaction and separation, and may pollute treated water. In contrast, nanofibers, which also have potential for PCA applications, have already proven favourable because of their recycling properties.17 One-dimensional (1D) nanostructures such as nanowires (NW), nanosheets (NS), nanorods (NR), and nanotubes (NT) enhance PCA due to their high surface areas.11,17,18 1D nanostructures are employed in various applications such as solving environmental (photocatalysis and water treatment)19,20 and energy (electronics and photonics) issues.21–23 In this context, photocatalysis is a ‘‘green” technique, which offers promising ways to completely remove toxic pollutants from the environment through its efficiency and broad suitability.24 1D nanostructures produced by electrospinning is straightforward and simple.25–27 Wang et al.28 reported that electrospun nanofibers of Bi2O3 exhibited significant PCA for degradation of the organic pollutant rhodamine B (RB). Furthermore, the Bi2O3 nanofibers could be easily recycled without decrease of PCA.
Herein, we report for the first time nanosheet-like structures from that of BiOCl and Bi2O3 electrospun nanofibers. The formation of these nanostructures and their morphology was observed at different precursor (BiOCl3 and Bi(NO3)3·5H2O) concentrations varying from 1 to 4 wt%. Microscopy, spectroscopy and XRD were used for the characterization of nanostructures. The resultant nanostructures were employed for the degradation of ARS dye under UV light (<390 nm) irradiation. The comparative PCA study of the electrospun nanostructures of how BiOCl and Bi2O3 induces complete degradation of the ARS dye was carried out. A plausible mechanism for the PCA of BiOCl is proposed and explained.
Experimental section
Materials
Bismuth(III) nitrate pentahydrate (Bi(NO3)3·5H2O); 98%, bismuth(III) chloride (BiCl3; 98%), polyacrylonitrile (PAN, MW = 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da), and N,N-dimethylformamide (DMF; anhydrous 99.8%) were obtained from Sigma Aldrich Chemical Company, Inc., St. Louis, MO, U.S. All chemicals were used without further purification.
000 Da), and N,N-dimethylformamide (DMF; anhydrous 99.8%) were obtained from Sigma Aldrich Chemical Company, Inc., St. Louis, MO, U.S. All chemicals were used without further purification.
Preparation
Bi2O3 nanofiber preparation. Nearly 10% (wt) of PAN powder was initially dissolved in DMF and stirred for about 2 h. After a reasonably miscible solution was obtained, nearly 1% w/v of Bi(NO3)3·5H2O was added to the above DMF solution, then stirred for about 2 h to create a homogeneous solution for the electrospinning. Similar solution preparations were repeated for the variable concentrations (2, 3, and 4 wt%) of Bi(NO3)3·5H2O. The procedure for the electrospinning was followed as published previously.25 The above electrospinning solution was drawn up into a 10 mL syringe with 21 G 1/2 gauge needle. The fiber deposition parameters (flow rate, applied voltage, drum rotation speed, etc.) were optimized until uniform nanofibers without bead formation was obtained. The flow rate was maintained at 0.5 mL h−1 (for Bi2O3) and 1.0 mL h−1 (for BiOCl). The distance between the electrodes (needle-tip and the collector) was maintained at about 10 cm. The applied electric voltage between the drum collector and the needle tip was 20 kV and the humidity level inside the electrospinning chamber was maintained at approximately 50%.
BiOCl nanofiber preparation. The BiOCl electrospinning solution is similar to that of the Bi2O3, except the precursor Bi(NO3)3·5H2O was replaced with BiCl3.The as-spun free standing nanostructures composed of uniform and continuous nanofibers were collected from the substrate. Later these nanostructures were vacuum dried to completely remove any residual solvents present on the surface. These nanofibers were then calcined at 500 °C for 5 h with a ramp rate of about 5 °C min−1 in air, and formed nanosheet-like structures.
Characterization
The surface characterizations of these structures were performed using field emission scanning electron microscopy (FE-SEM; Quanta 200F, FEI, Oregon, U.S.), prior to which the samples were coated with nearly 5 nm of gold (JEOL JFC-1200 fine coater, Japan) for better contrast. Transmission electron microscopy (TEM; JEOL JEM-2010F) at an acceleration voltage of 200 kV was used to characterize morphology. X-ray photoelectron spectroscopy (XPS; Kratos AXIS UltraDLD, Kratos Analytical Ltd., U.K.), with a mono Al Kα X-ray source (hν = 1486.71 eV) and operated at 15 kV and 5 mA, was used to identify the oxidation states of ions from the binding energies of photoelectrons. The pressure in the analysis chamber was maintained at 5 × 10−9 torr during each measurement.
Photocatalytic degradation
A suspension of the nanostructured photocatalyst with ARS aqueous solution was prepared in two steps. In the first step 5 mg of catalyst (BiOCl or Bi2O3) was dissolved in 45 mL of de-ionized (DI) water. In the second step 0.5 mg of ARS dye powder was dissolved in 5 mL of DI water. Then both the solutions were mixed and sonicated for about 30 min to obtain a good dispersion of solution. The initial concentration (C0) of the dye was noted before being exposed to the UV radiation. Later the dispersed solution was placed in such a way that it was completely surrounded by the light source (<390 nm). After every 10 min of irradiation, about 5 mL of the mixture was removed and centrifuged (Allegra 64R centrifuge, Beckman coulter) to remove the residues, and the UV spectra were measured for the supernatants (containing ARS).
Results and discussion
Surface characterization
As-spun nanofibers were characterized by FE-SEM and are shown in Fig. 1. It can be clearly seen that the nanofibers are randomly distributed throughout the area covered. The fibers spun from PAN/Bi(NO3)3·5H2O have smooth and uniform textured surfaces because of the amorphous nature of the PAN/Bi(NO3)3·5H2O (Fig. 1a–d). In contrast, the fibers from PAN/BiCl3 exhibit a rough textured morphology with continuous structures as shown in Fig. 1e–h. The average fiber diameters evaluated from the SEM images are 1 to 2 μm.
 |
| | Fig. 1 SEM images of as-spun nanofibers of Bi2O3 (a, b, c, d are 1, 2, 3 and 4% respectively) and BiOCl (e, f, g, and h are 1, 2, 3 and 4% respectively). | |
Upon calcination, the smoothly textured fiber morphology of Bi2O3 became converted to a porous morphology consisting of nanorods as shown in Fig. 2a–d. For all concentrations of Bi(NO3)3·5H2O, the nanostructures are porous due to shrinkage resulting from the decomposition of PAN at high temperatures. As seen in these SEM images, the fiber morphology is lost and the nanostructures become more like nanorod/nanotube-conjoined structures, which is due to the complete decomposition of the PAN polymer. The observed diameter of the nanostructures ranged from 40 nm to a few hundred nanometers. Formation of these elongated nanorods may be attributed to an anisotropic growth rate of the bismuth compound nanofibers. Huang, et al.29 reported that there is an increase in electrical potential for nanofibers with rougher surfaces. It has also been reported that the longer polymerization time for the nanofibers also led to the formation of polymer clusters and particles on the surface of nanofibers, making the surface rougher.30
 |
| | Fig. 2 SEM images after calcination of Bi2O3 (a, b, c, d are 1, 2, 3 and 4% respectively) and BiOCl (e, f, g, and h are 1, 2, 3 and 4% respectively). | |
Fig. 2e–h, shows the BiOCl nanostructures after calcination at about 500 °C for 5 hours. At the low BiOCl precursor concentrations (1, 2 wt%), the resulting structures as shown in Fig. 2e and f are not well defined and they overlap each other. But at high concentrations (3 and 4 wt%), as seen in Fig. 2g and h, the structures display nanosheets with anisotropic dimensions, and a unique nano-stack morphology with an average thickness and length of 700–800 nm and ∼2 μm respectively. These FE-SEM images show that the nanosheets appear to be located next to each other and that they are uniformly distributed within the scanning range. The lengths and widths of the nanosheets range up to about ∼12–15 μm. Fig. 3 shows the TEM images of the BiOCl and Bi2O3 nanostructures. Fig. 3a reveals that the Bi2O3 nanostructures are well-defined, relatively long particles with an average dimension of ∼200 nm. Bi2O3 is more vulnerable to electron-beam irradiation than is BiOCl, and hence Bi2O3 is very difficult to observe in high-resolution TEM images. This suggests that BiOCl would be the preferred compound for photocatalysis.
 |
| | Fig. 3 TEM images of nanosheets of (a) Bi2O3 and (b) BiOCl. | |
The structures of BiOCl and Bi2O3 were characterized by using powder X-ray diffraction (XRD) with CuKα (1.5418 Å) radiation as shown in Fig. 4. The XRD patterns are shown only for the highest level of doping (4 wt%) and after calcination at 500 °C. All of the XRD peaks have been identified and indexed. BiOCl belongs to the tetragonal space group P4/nmm according to the literature,31–33 whereas Bi2O3 exhibits the β-Bi2O3 phase (space group: P421c).34–36 The lattice parameters and crystallite sizes were calculated according to the Debye–Scherrer formula (D = kλ/βcosθ),37 where λ is the wavelength of the X-ray (1.5418 Å for CuKα), θ is the angle of diffraction, β is the full width at half maximum (FWHM) and k is the shape factor (=0.94 for spherical crystals with cubic symmetry). The calculated cell parameters are shown in Table 1. Since the volume of the Bi2O3 is larger than that of BiOCl, the photodecomposition of Bi2O3 may take a longer time. The crystal structure of BiOCl is composed of layers of Cl−, Bi+3 and O−2 ions and the ionic distances are dBi–Cl (3.06 Å) and dBi–O (2.32 Å).
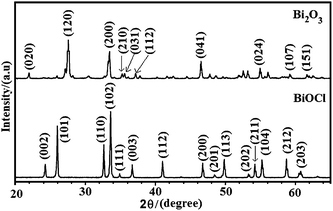 |
| | Fig. 4 Typical powder X-ray diffraction patterns of Bi2O3 and BiOCl after calcination at 500 °C, at highest concentrations (x = 4%). | |
Table 1 Calculated lattice parameter and crystallite sizes from XRD data. a, and c are the unit cell parameters
| |
Lattice parameters |
Cell volume (Å3) |
Crystallite size (Å) |
| a (Å) |
c (Å) |
| BiOCl |
3.8817 |
7.3682 |
111.02 |
2.96 |
| Bi2O3 |
7.7408 |
5.6338 |
337.57 |
2.95 |
XPS survey spectra of BiOCl and Bi2O3 are shown in Fig. 5. As demonstrated in Fig. 5, Bi, Cl, O and C are detected in the composite nanosheets by scanning high-resolution XPS spectra. The individual core levels of the nanostructures and their corresponding binding energies (BE) are presented in Fig. 6. The Bi4f of BiOCl, shown in Fig. 6a, comprises two asymmetric broad peaks, assigned to the Bi4f7/2(BE; 159.1 eV) and Bi4f5/2(BE; 164.2 eV). These two peaks were deconvoluted and the peaks at 164.8 and 160.0 eV are indexed as Bi+3 in BiOCl.38–41
 |
| | Fig. 5 XPS survey spectra of (A) Bi2O3 Bi2O3 (a, b, c, d are 1, 2, 3 and 4% respectively) and (B) BiOCl (e, f, g, and h are 1, 2, 3 and 4% respectively). | |
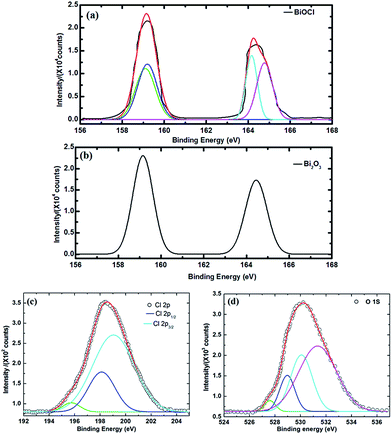 |
| | Fig. 6 High-resolution XPS spectra of (a) Bi4f in BiOCl; (b) Bi4f in Bi2O3; (c) Cl2p in BiOCl shows three contributions at 195.7, 198.1, and 199.0 eV, related to the defect band, Cl2p3/2 and Cl2p1/2 respectively; and (d) O1s core levels show bands at 527.6, 529.0, 530.1, and 531.3 eV. | |
The peaks at 164.1 and 159.1 for BiOCl can be indexed to lower Bi ionic states and are due to the oxygen vacancies present in the system.42 The appearance of the lower BE peaks might be due to the oxygen vacancies induced from the +3 ionic states.43–46 Essentially these same peaks, at 164.4 and 159.1 eV, and with the same intensity level, are observed for Bi2O3 as illustrated in Fig. 6b. The 4f7/2 and 4f5/2 orbitals were formed from the bismuth oxide species during the calcination process. However, the energy differences between 4f7/2 and 4f5/2 are ∼5.1 eV for BiOCl and 5.3 eV for Bi2O3, which are approximately the same and which indicates that the main valence state of Bi in both nanostructures is Bi+3.40,47 The Cl2p spectra for BiOCl is shown in Fig. 6c. There is a broad 2p peak, which can be decomposed to triplet peaks at 198.1 and 199.0 eV, and which are assigned to Cl2p3/2 and Cl2p1/2, respectively. The ∼0.9 eV energy difference between these two peaks is a characteristic feature for Cl− anions. This result further confirms the presence of chlorine atoms in the BiOCl fibre nanostructures. Another peak at 195.7 eV corresponds to a defect peak. As for the O1s spectrum shown in Fig. 6d, a broad peak is identified and the shape of this peak indicates that there can be more than one chemical state according to the binding energy. This peak is decomposed at the binding energies of 527.6, 529.0, 530.1, and 531.3 eV for both nanostructures.48,49 The increased binding energies are simply related to the crystal lattice oxygen (OBi–O), surface hydroxyl groups (OOH), and adsorbed water. The peak at 529.0 eV is assigned to oxygen bound to trivalent Bi ions and the small shoulder at 531.3 eV implies that the surface is partially covered with hydroxide OH groups.50 The binding energies related to carbon (C1s) for BiOCl and Bi2O3 are 284.8 and 284.6 eV respectively, as presented in Fig. SI1 (ESI),† and are due to a surface layer of carbon contamination. The C1s peak of PAN at 286.5 eV is a related carbon species that has not been identified, implying that the organic species has been degraded completely51 during calcination at 500 °C.
Photocatalysis and photodegradation
To study the photodegradation, UV-irradiated ARS dye solutions with and without catalyst need to be investigated. And, in the present studies, the PCA properties of ARS dye with and without photocatalysts have been determined under UV-light irradiation. The corresponding photocatalysis studies after incorporation of BiOCl and Bi2O3 nanostructures were analyzed and illustrated in Fig. SI2 and SI3.† The ARS solution was red with and without the addition of catalyst nanostructures. But while decolourization was not observed without catalyst, a change in colour was observed after adding the catalysts. These observations indicate that there is no significant degradation and decolorization of ARS dye in the absence of the photocatalyst. Upon UV irradiation of the ARS solution containing the catalysts, degradation was caused and observed as decolourisation of the reaction solution from red to pale pink or white. BiOCl performed better than did Bi2O3. The superior degradation performance of BiOCl was due to the difference in sizes of the catalysts as this also influences the adsorption of the dye molecules.
The related degradation curves with varying concentrations of the precursor are shown in Fig. 7, and the rates of degradation calculated from these curves are tabulated in Table 2. Bi2O3 at a concentration of 2% caused significantly faster PCA than it did at any of the other three concentrations. The faster PCA may be because of the morphology change: nanofibers were converted to highly porous nanorods at that particular concentration, as seen in Fig. 2b. BiOCl shows a PCA enhancement at a concentration of 3%, where the nanosheet-like structures were formed (see Fig. 2g).
 |
| | Fig. 7 Photocatalytic ARS dye degradation of (a) Bi2O3 and (b) BiOCl under UV-irradiation. | |
Table 2 Kinetic constants and their calculated half-life time in minutesa
| |
x = 1% |
x = 2% |
x = 3% |
x = 4% |
| k |
t1/2 |
k |
t1/2 |
k |
t1/2 |
k |
t1/2 |
| x represents the concentrations, k is the kinetic constant and t1/2 is the half-life of the catalyst. |
| Bi2O3 |
0.124 |
5.584 |
0.165 |
4.141 |
0.141 |
4.933 |
0.136 |
5.106 |
| BiOCl |
0.133 |
5.207 |
0.152 |
3.530 |
0.196 |
4.568 |
0.138 |
5.036 |
The photoreaction curves for Bi2O3 were also plotted and are shown in Fig. 7a, The photodegradation rate (C/C0) of the dye with different concentrations (1, 2, 3, and 4%) of precursor were observed under UV light irradiation within the marked time spans, where ‘C0’ is the initial concentration of the dye at time T = ‘0’, and ‘C’ is the final concentration at time T = ‘t’. From the observations of the absorption spectrum (SI2†), the degradation is over 75%, indicating excellent photocatalytic efficiency (η%). The efficiency was high, at 76.53%, with a Bi2O3 precursor concentration of 2%, where the morphology is quite different than for the other concentrations. The other concentrations also showed relatively good photocatalytic efficiencies within 130 min (73.14, 73.72, and 75.38% for 1, 3, and 4% respectively). As the concentration of the catalyst was initially increased, the photocatalytic rate efficiency reached an optimum, and then started to come down with additional increases in concentration. Fig. 7b, shows the photocatalytic degradation of BiOCl and an aqueous solution of ARS under UV-light illumination. It can be noted that the absorption band of the dye degraded rapidly at the precursor concentration level of 3% and complete degradation happened in 70 min. For the other concentrations (1, 2 and 4%), the degradation process went on for 80 min. The rapid photodegradation rate as shown in Fig. 7b is probably because of the ordered morphology of the nanosheets, whereas at the first two concentrations (1 and 2%), no clear morphology could be seen in the SEM images (Fig. 2e and f). The highest efficiency was found at the concentration x = 3%, the value being 99.34%. The other concentrations (1, 2 and 4%) also showed relatively good photocatalytic efficiencies, being a little lower at 88.54, 96.94, and 92.34% for x = 1, 2, and 4% respectively.
The kinetics plots of Bi2O3, and BiOCl are presented in Fig. 8 and 9. Both of the nanostructures follow pseudo first-order kinetics. From the kinetic plots, the rate constants were determined by linear polynomial fitting of the ln(C/C0) curve and the rate constant ‘k’ values were evaluated from the equation, k = 2.303 × log(C/C0) and presented in Table 2. The rate constant at 2% and 3% dopings of Bi2O3 and BiOCl shows the best degradation performance, since at those concentrations the morphology of the catalysts was quite different.
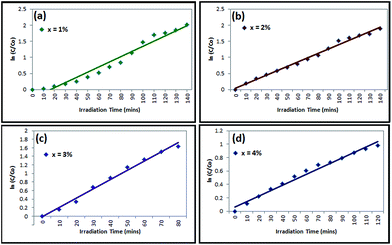 |
| | Fig. 8 Kinetic plots and rate constant evaluation of Bi2O3 photocatalyst at (a) x = 1%, (b) x = 2%, (c) x = 3% and (d) x = 4% (w/v). | |
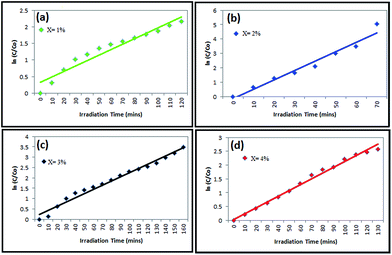 |
| | Fig. 9 Kinetic plots and rate constant evaluation of BiOCl photocatalyst at (a) x = 1%, (b) x = 2%, (c) x = 3% and (d) x = 4% (w/v). | |
From pseudo first-order kinetic constants, the half-life time (t1/2) can be calculated using the expression t1/2 = 0.693/k. According to the half-life expression, the half-life of the dye degradation has been evaluated and tabulated in Table 2. It also suggests that the BiOCl is an excellent photocatalyst for the industrial dye degradation, and is superior to Bi2O3.
Dye degradation mechanism
The BiOCl nanosheet-like structures showed significant PCA enhancement in the decomposition of ARS dye. In general, the PCA of the catalyst is dependent on several factors such as surface properties, crystallinity, morphology, optical properties, and diameter.52 From XRD analysis, crystal structures were not changed much, which had minimal effect in PCA performance. It is worth noting that the morphology of BiOCl changed into nanosheet-like structures that might adsorb more dye molecules at the surface. The PCA reaction might be preceded by two steps:32,53 adsorption, and degradation after the adsorption. Thus, the stronger the adsorption capability, the better would be the subsequent photodegradation.
The possible mechanism for ARS dye degradation in the presence of BiOCl catalyst has been proposed and is presented in Fig. 10. The conduction band (CB) and valance band (VB) potentials of BiOCl are −1.10 and −2.20 eV respectively,32 and the band gap is hence 3.3 eV.1,54 The ARS dye is adsorbed on the surface of the catalyst (BiOCl). Photogenerated electrons transfer from VB to CB, leaving holes behind VB atop of the ARS dye. Then the excited electrons react with the oxygen to form O2−. Then these oxygen radicals and holes can effectively oxidize the ARS dye. Thus, two mechanisms may be involved in the degradation.25
| | |
B(catalyst) + hv → h+ + e−
| (1) |
| | |
e− + O2→ ˙O2− (radical)
| (2) |
| | |
h+ + OH− → ˙OH (radical)
| (3) |
| | |
ARS*(dye.ads) + B(catalyst) → B(catalyst)(e−) + ARS(dye.ads)+
| (4) |
| | |
B(catalyst)(e−) + O2 → B(catalyst) + ˙O2− (radical)
| (5) |
| | |
B(catalyst) + ˙O2− + 2H+ → B(catalyst) + ˙OH (radical)
| (6) |
| | |
ARS(dye.ads) + ˙O2− → Products
| (7) |
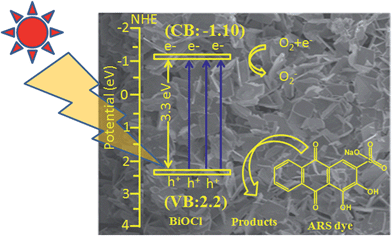 |
| | Fig. 10 Schematic representation of oxidation and reduction in BiOCl. | |
In eqn (1)–(7), B stands for BiOCl, and dye.ads stands for adsorbed dye. These equations provide mechanisms for photoadsorption and consequent dye degradation.
Conclusions
Electrospun sheet/rod-shaped nanostructures have been produced. By changing the precursor concentration, changes in the morphology of Bi2O3 and BiOCl nanostructures have been observed from SEM, and TEM confirmed their morphologies. During calcination, the dynamics of the polymers vary and the collective interaction between the precursors causes anisotropic crystal formation. XRD reveals the tetragonal and β-phases of BiOCl and Bi2O3 respectively. From XPS, it is confirmed that Bi in these structures exists in the Bi+3 ionic state, and in lower ionic states as well. The converted nanostructures are employed for the photodegradation of ARS dye. As the size of the crystallite increases (determined from XRD), the photodecomposition ability substantially decreases. Therefore, the enhancement of PCA is due to the change in morphology, such as that into nanosheet-like structures. Possible degradation mechanisms for the dye have been proposed and discussed. We anticipate that our investigations may help further understanding of various nanostructured materials and accelerate their potential applications.
Acknowledgements
All the authors thank M3TC (EDB) R-261-501-018-414, Singapore for providing financial assistance. V J Babu thanks The Scientific & Technological Research Council of Turkey (TUBITAK) (TUBITAK-BIDEB 2221, Fellowships for visiting scientists and scientists on sabbatical leave) for providing fellowship support.
Notes and references
- H. Cheng, B. Huang and Y. Dai, Nanoscale, 2014, 6, 2009–2026 RSC.
- K.-L. Zhang, C.-M. Liu, F.-Q. Huang, C. Zheng and W.-D. Wang, Appl. Catal., B, 2006, 68, 125 CrossRef CAS PubMed.
- Y. Bessekhouad, D. Robert and J.-V. Weber, Catal. Today, 2005, 101, 315–321 CrossRef CAS PubMed.
- W. Xiaohong, Q. Wei and H. Weidong, J. Mol. Catal. A: Chem., 2007, 261, 167–171 CrossRef PubMed.
- L. Zhang, W. Wang, J. Yang, Z. Chen, W. Zhang, L. Zhou and S. Liu, Appl. Catal., A, 2006, 308, 105–110 CrossRef CAS PubMed.
- H. Peng, C. K. Chan, S. Meister, X. F. Zhang and Y. Cui, Chem. Mater., 2009, 21, 247–252 CrossRef CAS.
- G. G. Briand and N. Burford, Chem. Rev., 1999, 99, 2601–2658 CrossRef CAS PubMed.
- S. K. Poznyak and A. I. Kulak, Electrochim. Acta, 1990, 35, 1941–1947 CrossRef CAS.
- D. O. Charkin, P. S. Berdonosov, V. A. Dolgikh and P. Lightfoot, J. Solid State Chem., 2003, 175, 316–321 CrossRef CAS.
- N. Kijima, K. Matano, M. Saito, T. Oikawa, T. Konishi, H. Yasuda, T. Sato and Y. Yoshimura, Appl. Catal., A, 2001, 206, 237–244 CrossRef CAS.
- X. Zhang, Z. Ai, F. Jia and L. Zhang, J. Phys. Chem. C, 2008, 112, 747–753 CAS.
- W. Xiaohong, Q. Wei and H. Weidong, J. Mol. Catal. A: Chem., 2007, 261, 167–171 CrossRef PubMed.
- X. Lin, T. Huang, F. Huang, W. Wang and J. Shi, J. Phys. Chem. B, 2006, 110, 24629–24634 CrossRef CAS PubMed.
- L. Zhang, J. Li, Z. Chen, Y. Tang and Y. Yu, Appl. Catal., A, 2006, 299, 292–297 CrossRef CAS PubMed.
- J. C. Yu, J. Yu, W. Ho and L. Zhang, Chem. Commun., 2001, 1942–1943 RSC.
- T. Park, S. A. Haque, R. J. Potter, A. B. Holmes and J. R. Durrant, Chem. Commun., 2003, 2878–2879 RSC.
- Z. Liu, D. D. Sun, P. Guo and J. O. Leckie, Nano Lett., 2007, 7, 1081–1085 CrossRef CAS PubMed.
- Y. Wu, H. Yan and P. Yang, Top. Catal., 2002, 19, 197–202 CrossRef CAS.
- J. Zhu and M. Zäch, Curr. Opin. Colloid Interface Sci., 2009, 14, 260–269 CrossRef CAS PubMed.
- V. J. Babu, A. S. Nair, Z. Peining and S. Ramakrishna, Mater. Lett., 2011, 65, 3064–3068 CrossRef CAS PubMed.
- X. Hu, G. Li and J. C. Yu, Langmuir, 2010, 26, 3031–3039 CrossRef CAS PubMed.
- P. Balaya, Energy Environ. Sci., 2008, 1, 645–654 CAS.
- V. J. Babu, D. V. B. Murthy, V. Subramanian, V. R. K. Murthy, T. S. Natarajan and S. Ramakrishna, J. Appl. Phys., 2011, 109, 074306 CrossRef PubMed.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev, 1995, 95, 69 CrossRef CAS.
- J. B. Veluru, S. R. S. Bhavatharini and S. Ramakrishna, RSC Adv., 2014, 4, 19251–19256 RSC.
- J. B. Veluru, K. K. Manippady, M. Rajendiren, K. M. Mya, P. R. Rayavarapu, S. N. Appukuttan and R. Seeram, Int. J. Hydrogen Energy, 2013, 38, 4324–4333 CrossRef CAS PubMed.
- V. J. Babu, M. K. Kumar, A. S. Nair, T. L. Kheng, S. I. Allakhverdiev and S. Ramakrishna, Int. J. Hydrogen Energy, 2012, 37, 8897–8904 CrossRef CAS PubMed.
- C. Wang, C. Shao, L. Wang, L. Zhang, X. Li and Y. Liu, J. Colloid. Interface Sci., 2009, 333, 242–248 CrossRef CAS PubMed.
- Z. M. Huang, Y. Z. Zhang, M. Kotaki and S. Ramakrishna, Compos. Sci. Technol., 2003, 63, 2223–2253 CrossRef CAS.
- Z. Wang, Z.-Z. Zhu, J. Shi and H.-L. Li, Appl. Surf. Sci., 2007, 253, 8811–8817 CrossRef CAS PubMed.
- L. Q. Ye, L. Zan, L. H. Tian, T. Y. Peng and J. J. Zhang, Chem. Commun., 2011, 47, 695–697 RSC.
- T. Xie, L. Xu, C. Liu, J. Yang and A. M. Wang, Dalton Trans., 2014, 43, 2211–2220 RSC.
- H. Liu, W. Yang, Y. Ma and J. Yao, Appl. Catal., A, 2006, 299, 218–223 CrossRef CAS PubMed.
- B. Gao, Y. J. Kim, A. K. Chakraborty and W. I. Lee, Appl. Catal., B, 2008, 83, 202 CrossRef CAS PubMed.
- M. Gotić, S. Popović and S. Musić, Mater. Lett., 2007, 61, 709–714 CrossRef PubMed.
- H. Cheng, B. Huang, J. Lu, Z. Wang, B. Xu, X. Qin, X. Zhang and Y. Dai, Phys. Chem. Chem. Phys., 2010, 12, 15468–15475 RSC.
- J. I. Langford and A. J. C. Wilson, J. Appl. Crystallogr., 1978, 11, 102–113 CrossRef CAS.
- H. An, Y. Du, T. Wang, C. Wang, W. Hao and J. Zhang, Rare Met., 2008, 27, 243–250 CrossRef CAS.
- Y. Wu, M. Xing, J. Zhang and F. Chen, Appl. Catal., B, 2010, 97, 182–189 CrossRef CAS PubMed.
- W. E. Morgan, W. J. Stec and J. R. V. Wazer, Inorg. Chem., 1973, 12, 953–955 CrossRef CAS.
- J.-M. Song, C.-J. Mao, H.-L. Niu, Y.-H. Shen and S.-Y. Zhang, CrystEngComm, 2010, 12, 3875–3881 RSC.
- L. Ye, K. Deng, F. Xu, L. Tian, T. Peng and L. Zan, Phys. Chem. Chem. Phys., 2012, 14, 82–85 RSC.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- M. Batzill, E. H. Morales and U. Diebold, Chem. Phys., 2007, 339, 36–43 CrossRef CAS PubMed.
- C. Rath, P. Mohanty, A. C. Pandey and N. C. Mishra, J. Phys. D: Appl. Phys., 2009, 42, 205101 CrossRef.
- G. Liu, H. G. Yang, X. Wang, L. Cheng, H. Lu, L. Wang, G. Q. M. Lu and H.-M. Cheng, J. Phys. Chem. C, 2009, 113, 21784–21788 CAS.
- V. S. Dharmadhikari, S. R. Sainkar, S. Badrinarayan and A. Goswami, J. Electron Spectrosc. Relat. Phenom., 1982, 25, 181–189 CrossRef CAS.
- http://www.lasurface.com/database/elementxps.php.
- http://srdata.nist.gov/xps/selEnergyType.aspx.
- C. Wang, C. Shao, X. Zhang and Y. Liu, Inorg. Chem., 2009, 48, 7261–7268 CrossRef CAS PubMed.
- C. Wang, C. Shao, Y. Liu and L. Zhang, Scr. Mater., 2008, 59, 332–335 CrossRef CAS PubMed.
- I.-S. Cho, S. Lee, J. H. Noh, G. K. Choi, H. S. Jung, D. W. Kim and K. S. Hong, J. Phys. Chem. C, 2008, 112, 18393–18398 CAS.
- J. Di, J. Xia, S. Yin, H. Xu, L. Xu, Y. Xu, M. He and A. H. Li, RSC Adv., 2014, 4, 14281–14290 RSC.
- L.-P. Zhu, G.-H. Liao, N.-C. Bing, L.-L. Wang, Y. Yang and H.-Y. Xie, CrystEngComm, 2010, 12, 3791–3796 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional UV photocatalytic ARS dye degradation studies for the Bi2O3 and BiOCl, being provided. See DOI: 10.1039/c4ra03754e |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 








