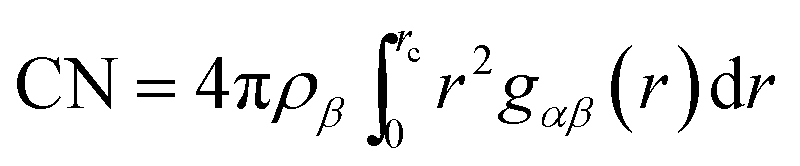DOI:
10.1039/C4RA03678F
(Paper)
RSC Adv., 2014,
4, 34267-34280
Effects of the temperature and trehalose concentration on the hydrophobic interactions of a small nonpolar neopentane solute: a molecular dynamics simulation study†
Received
23rd April 2014
, Accepted 16th July 2014
First published on 16th July 2014
Abstract
The influence of trehalose on the temperature-induced changes of hydrophobic interactions is investigated employing classical molecular dynamics simulations. The neopentane molecules are used to model typical nonpolar entities. Six different trehalose concentrations are used and for each concentration five different temperatures are considered. The neopentane–neopentane potentials of mean force (PMF), selected site–site distribution functions involving different solution species, the hydrogen bond properties and trehalose induced modifications of the translational diffusion coefficients of solution species are reported. The calculations of PMF followed by estimation of the association constant values suggest that the trehalose induced reduction in the hydrophobic interactions observed at low temperature is getting reduced at higher temperature. This finding is further supported by calculating the neopentane–water distribution functions. Our calculations of the hydrogen bond properties reveal that in contrast to a pure water system, the average number of water–water hydrogen bonds in a concentrated trehalose solution is insensitive to the temperature change. The analyses of trehalose cluster structures reveal the formation of a higher order trehalose cluster on increasing the trehalose concentration and the breaking of these higher order clusters as temperature increases. With increasing the trehalose concentration, we observe retardation of the translational motion of all solution species and this slowing down of the translational dynamics is more pronounced for trehalose than for water.
I. Introduction
The biological activity of a protein in living systems depends on its three-dimensional native structure. Under very different extreme conditions such as desiccation, high pressure, low and high temperature. etc., proteins may denature and thereby lose their activity.1 Living systems adopt some techniques to protect proteins in such extreme conditions.2 The synthesis of sugar is a good example of the defensive reaction of many organisms. It has been found that the aqueous solutions of simple carbohydrates protect the proteins and helps in retaining the proteins' activity.3,4 Among all carbohydrates, trehalose has often been depicted as the most effective protector of biomolecules for its very interesting bioprotecting abilities.1,5 Trehalose, a nonreducing disaccharide, is composed of two glucopyranosyl units connected together through an α,α-(1 → 1) glycosidic oxygen linkage between their anomeric carbon atoms (see Fig. 1). Because of its bioprotecting ability, trehalose has received huge attention in a wide range of applications, e.g., for long-term preservation of therapeutic proteins, foods and cosmetics in industry.6–8 Further, as reported recently, it can also act as a stabilizer to stabilize blood platelet cells.9 It can be found in a wide variety of organisms such as bacteria, fungi, insects, invertebrates as well as in some flowering plants and vascular plants.10
 |
| | Fig. 1 Trehalose molecule. All carbons, oxygens and hydroxyl hydrogens are labeled. The other hydrogen atoms are left out for clarity. | |
To understand the trehalose's role as a structure protecting molecule, three hypotheses have been proposed for: (1) mechanical entrapment: trehalose molecules form a highly viscous glassy matrix that protects the biomolecule in its biological conformation (due to strong reduction in the protein dynamical fluctuations) like an insect trapped in amber.11,12 (2) Water replacement hypothesis: this hypothesis suggests that trehalose molecules replace water molecules from a protein hydration shell (and form direct hydrogen bonds with the protein) thereby stabilizing the protein three-dimensional structure during dehydration.13–15 (3) Water entrapment hypothesis: according to this hypothesis trehalose molecules do not bind directly to biomolecules. Rather, they entrap the water molecules in the intermediate layer between them and the biomolecule, thus preserving the native solvation of the biomolecule.16 This mechanism indirectly suggests the preferential exclusion of trehalose molecules from the protein surface17,18 and acts as preferential hydration hypothesis of the biomolecule. Though widely studied,15,19–25 the origin and mechanisms of the trehalose's bioprotecting ability are yet to be understood fully and none of the above hypotheses are considered as completely accepted. As a result, it remains a subject of active research. For example, the formation of a direct sugar–protein hydrogen bond has been reported in IR spectra studies of a dry protein–sugar mixture supporting the water replacement hypothesis.15,25 Another Raman spectroscopy study reported trehalose induced retention of the protein hydration layer even in a low water content, and the presence of trapped water molecules in the glassy matrix formed by trehalose molecules. As a result, the protein retains its native structure and biological activity. This observation is in accordance with the water entrapment hypothesis.16
The hydrophobic and hydrogen bond interactions within the secondary and tertiary structures contribute to the thermal stability of proteins. Hydrogen bonding and hydrophobic interactions are two main driving forces of protein folding–unfolding process. Though a recent molecular dynamics simulation study argues about the role of the hydrogen bond on protein conformation,26 other studies reveal that the contribution of hydrogen bonding interactions is 1.4 kcal mol−1 per hydrogen bond and for hydrophobic interactions it is 1.2 kcal mol−1 per CH2 group.27,28 Nevertheless, these values suggest that the protein stability is sensitive to changes in the environmental and chemical conditions such as temperature, pressure, presence of denaturants (e.g., urea and guanidium hydrochloride), salts, etc. In proteins, since the backbone atoms are identical, the main structural difference among the proteins arises due to the presence of different side chains.29–31 These suggest that the hydrophobic interactions among different side chains play an important role in stabilizing the protein native state. Since the interactions between different groups of protein and different solvent and cosolvent molecules are very complex, in this study, mimicking the protein interior by simple hydrophobic neopentane molecules, we try to reduce the complexity of highly interacting systems present in the actual protein–sugar solution. The choice of hydrophobic neopentane molecules as solutes over the vastly used methane is due to the fact that neopentane breaks the hydrogen bond network of water whereas methane fits into it. Moreover, the effective size of the nonpolar side chains in the protein is larger than the size of a methane molecule. As mentioned above, since trehalose molecules act as a biomolecule stabilizer at high and low temperatures, in the present study, our focus is mainly on the role of trehalose on the neopentane hydrophobic interactions at various temperatures. Since our model neopentane solute does not possess any hydrophilic (hydrogen bonding) site, the solute–solvent hydrogen bonding is missing from the present study which is essential for suggesting the bioprotective mechanism of trehalose completely. We have considered a wide range of temperatures (from 285 to 345 K) and our goal is to see how temperature induced changes in the hydrophobic interactions are getting affected in the presence of trehalose and thereby predict the role of trehalose in protein stability at different thermal conditions. In this article, we therefore describe the solvation characteristics of hydrophobic neopentane solutes in aqueous trehalose solutions of varying concentrations. By calculating the potentials of the mean force (PMFs) followed by estimation of the association constant value, we first examine whether trehalose has any significant effect on the temperature dependent changes in the neopentane aggregation in different solutions. An insight picture of the effect of trehalose on the neopentane aggregation is obtained by examining its effects on the water structure and also its direct interactions with the hydrophobic neopentane molecules. We use the familiar water oxygen–oxygen radial distribution function (rdf) to describe water structuring. Further, the solute–solvent coordination number is used to calculate the preferential binding with the solute. In the second part of this article, we concentrate on trehalose and temperature induced changes in the hydrogen bond properties and on the translational dynamics of different solution species.
The remainder of this paper is organized into three parts. The model and simulation details are briefly described in section II, the results are presented and discussed in section III, and our conclusions are briefly summarized in section IV.
II. Models and simulation method
Classical molecular dynamics simulations of neopentane–trehalose–water mixtures were carried out with six different trehalose concentrations and at five different temperatures. We employed a GLYCAM06 force field for trehalose32 and the SPC/E33 potential was used for water. For neopentane we used the OPLS/AA34 potential because of the fact that compared to a single site united atom model the aggregation propensity of neopentane in water increases as the branching increases. As already reported, the association of neopentane molecules is greater for a branched five-site model than a single-site neopentane model.35 Furthermore, the OPLS/AA neopentane model shows the characteristics one would expect from a large hydrophobic surface.36 Since our goal is to explore the effect of trehalose on neopentane–neopentane hydrophobic interactions, we consider the OPLS/AA neopentane model. We use the AMBER10 software for all simulations.37 We note that a comparative study between different disaccharide force fields shows that the results of the GLYCAM06 force field (which is mostly used for simulations of monosaccharides and oligosaccharides) are in good agreement with the DFT and experimental values.38,39 A comparison between different water models reveals that the structural and dynamical properties of SPC/E water match well with the experimental results.40 In this regard, we further note that a combination of a GLYCAM06 force field (for trehalose and kojibose) and SPC/E water has recently been used elsewhere.41 The systems considered here are summarized in Table 1. The initial configurations of our systems were prepared using the Packmol program.42 All simulations were carried out in a cubic box with a temperature range of 285 to 345 K. In order to obtain a reasonable initial structure, the systems were energy minimized for 5000 steps, where the first 2500 steps in the steepest descent method were followed by the same number of steps in the conjugate gradient method. After that each system was heated up slowly from 0 K to the desired temperature for 100 ps in a canonical ensemble (NVT) and then the systems were equilibrated in an isothermal–isobaric (NPT) ensemble for 5 ns. For calculating the different structural and hydrogen bond properties, the production runs were performed for 35 ns in an isothermal–isobaric (NPT) ensemble and at 1 atm pressure. To maintain physical pressure, a Berendsen thermostat with a pressure relaxation time of 2 ps was used43 and the temperature was controlled by the Langevin dynamics method with a collision frequency of 1 ps−1. In all simulations, periodic boundary conditions were applied to remove the edge effects and a time step of 2 fs was used. A cutoff distance of 10 Å was considered for all non-bonding interactions and the long-range electrostatic interactions were treated using the particle mesh Ewald method. Bonds involving hydrogen were constrained by applying the SHAKE algorithm.44 For calculating the diffusion coefficients of different solution species, the simulations were continued for another 15 ns in a microcanonical (NVE) ensemble. All simulations were carried out on a High Performance Computer Cluster having a dual socket (8 core × 2 processor) Intel Sandybridge E5-2670 processor running at 2.6 GHz and a Linux based (Cent OS 6.2) operating system. Finally, the trajectories generated from the production run were analyzed using the ptraj program of the AMBER10 toolkit and the Visual Molecular Dynamics (VMD) package.45
Table 1 Nneo, Ntre, Nwat, volume and wtre represent the number of neopentane, trehalose, water molecules, box volumes and weight percentage of trehalose, respectively, for different systems
| System |
Nneo |
Ntre |
Nwat |
Volume (nm3) |
| 285 K |
300 K |
315 K |
330 K |
345 K |
wtre% |
| S0 |
20 |
0 |
1400 |
45.30 |
45.75 |
45.88 |
46.58 |
47.32 |
0 |
| S1 |
20 |
10 |
1400 |
48.84 |
49.10 |
49.91 |
50.16 |
50.88 |
11 |
| S2 |
20 |
20 |
1400 |
52.20 |
52.43 |
52.95 |
54.01 |
54.25 |
20 |
| S3 |
20 |
50 |
1400 |
63.00 |
63.14 |
63.74 |
64.72 |
64.98 |
39 |
| S4 |
20 |
75 |
1400 |
68.75 |
68.92 |
72.49 |
73.51 |
74.34 |
49 |
| S5 |
20 |
100 |
1400 |
79.80 |
80.78 |
88.66 |
82.71 |
83.47 |
56 |
III. Results and discussion
A. Neopentane–neopentane pair potentials of the mean force
In order to get a picture of the hydrophobic interaction at a pair level, we have calculated the potentials of the mean force of hydrophobic neopentane solutes from the solute–solute pair correlation function (gss(r)) using the relation| |
W(r) = −kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lngss(r) lngss(r)
| (1) |
where kB is the Boltzmann constant, T is the absolute temperature and the pair correlations function (g(r)) is the ratio of probability of finding a pair of atomic sites at a distance r and the probability expected for a completely random distribution at the same density. Note that in order to calculate W(r), we have considered a neopentane–neopentane pair correlation function involving neopentane central carbon atoms.
The PMF profiles of neopentane in aqueous solutions of varying trehalose concentrations at five different temperatures are shown in Fig. 2. In each PMF curve, we observe two minima separated by a maximum. The first minimum at about 5.85 Å that arises from the direct contact of two neopentane molecules, is commonly known as the contact minimum (CM) and the relatively weaker second minimum that appears at about 10.25 Å is ascribed to the solvent-separated minimum (SSM). The presence of these two minima reveals the existence of the associated and solvent-separated states of neopentane. The maximum between the two minima that appears at about 8.5 Å is known as the dissolvation barrier (BARR).
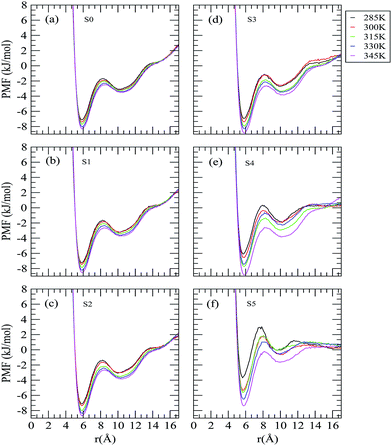 |
| | Fig. 2 Neopentane–neopentane potential of the mean forces for different systems. | |
It is apparent from Fig. 2 that the well depth of CM is higher than that of SSM. Considering a system without trehalose (system S0) first, we find that the small system size of our simulation and the finite neopentane aggregates make the PMF profile have a positive gradient that does not go to zero at large solute separations.46 With regard to the effect of increased temperature on the PMF profiles, we observe: (a) a slightly outward movement of CM, SSM and BARR; (b) an enhancement in the well depth of both CM and SSM, indicating more pronounced free energy basins of both contact and solvent-separated states of neopentane; and (c) no change in the positive gradients observed at higher solute separation. At a given temperature, with regard to the influence of the trehalose concentration on the PMF of neopentane, two points are worth noting: (a) reduction in the well depths of both CM and SSM with increasing trehalose concentrations and the effect is more pronounced at lower temperatures; and (b) reduction in the slope of PMF curves at larger solute separation with increasing trehalose concentration. The PMF difference between SSM and CM, (ΔWf→u) which indicates the association tendency of the hydrophobic neopentane molecules, is related to the thermodynamic stability of the neopentane associated state. For example, for system S0 at 285 K, the value of ΔWf→u is +4.03 kJ mol−1 indicating that a neopentane molecule prefers to be in contact with other neopentane molecules rather than to be separated by solvent water molecules. Fig. 3 displays the variations in the ΔWf→u values with changing trehalose concentration at different temperatures. As can be seen, in comparison to low temperature, trehalose helps stabilize the contact state at high temperatures when the trehalose concentration is also high. Interestingly, high trehalose concentration diminishes the hydrophobic interactions between the neopentane molecules at 285 K. These findings suggest that trehalose induced changes in the hydrophobic interactions between the neopentane molecules are influenced dramatically by changes in the temperature.
 |
| | Fig. 3 Free energy differences between SSM and CM for different systems. | |
For better understanding of the effect of increased temperature on the neopentane aggregation for various trehalose concentrations, we have calculated the neopentane–neopentane association constant, Ka, by integrating the PMF to the first maximum (the position of the barrier), which defines the outer limit of the solute–solute contact configuration. Ka is defined as47,48
| |
 | (2) |
where
ra is the position of the barrier in the corresponding PMF curve. Note that the higher the value of
Ka, the greater will be the association tendency of the hydrophobic moieties in the aqueous system. Any perturbation that favors the associated state of the solute will, therefore, increase the value of
Ka. The values of
Ka for different systems are presented in
Table 2. It is apparent that for systems S0 and S1, the value of the association constant is very large and with increasing temperature it remains practically unchanged. Interestingly, for other systems, the value of
Ka increases with increasing temperature and this effect is more pronounced for systems of higher trehalose concentrations (
i.e., systems S4 and S5). For example, for system S5, there is a three fold increase in the
Ka value as temperature is increased from 285 to 345 K. Furthermore, for a particular temperature, with increasing trehalose concentration the association constant value decreases. Though the decrease in its value is more pronounced at lower temperature, remarkably, at higher temperature the value of
Ka changes noticeably only at higher trehalose concentrations. These observations suggest that the trehalose induced depletion in the neopentane–neopentane hydrophobic interaction observed at low temperature, is getting reduced at high temperature and the trehalose molecules act as a stabilizing co-solute for hydrophobic interactions at high temperature. Here we note that, for system S0, where there is no trehalose molecule, the temperature has essentially no influence on the neopentane association constant value. This finding is in contradiction to the results of the molecular dynamics simulation study of methane-like molecules in pure water reported by Lüdemann
et al.49 They reported that as temperature increases, the equilibrium between the contact pair and the solvent-separated pair is shifted towards the former. In this regard, it is also worth noting that for system S0, the temperature induced changes in Δ
Wf→u observed here (see
Fig. 3) are due to the presence of the temperature factor in the PMF expression.
Table 2 Association constant, Ka (M−1), of neopentane in different systems
| System |
S0 |
S1 |
S2 |
S3 |
S4 |
S5 |
| 285 K |
8.26 |
8.54 |
7.94 |
7.18 |
4.42 |
1.42 |
| 300 K |
8.34 |
8.31 |
7.80 |
7.40 |
5.12 |
2.83 |
| 315 K |
8.30 |
8.55 |
9.05 |
8.44 |
7.46 |
2.90 |
| 330 K |
8.41 |
8.54 |
9.17 |
8.57 |
5.98 |
3.75 |
| 345 K |
8.47 |
8.93 |
9.37 |
9.66 |
9.29 |
4.98 |
B. Neopentane cluster structure analysis
In order to quantify the clustering of hydrophobic neopentane molecules and the possible effects of trehalose and temperature on it, we have estimated the average cluster size in all systems simulated here. For this, we have adopted the method employed by Martinez et al.50 for a Lennard-Jones fluid. Our group in the simulation study of methane aggregation in aqueous osmolyte solutions successfully employed this method later.48 Each neopentane molecule can be assigned to low-, average-, or high-density clusters with like molecules according to the following relations:
| average, if n0 − δ ≤ n ≤ n0 + δ |
Note that, in order to calculate the number of molecules in the solvation sphere (n) we have integrated the neopentane–neopentane pair correlation function up to the first minimum. And for calculating the average number of molecules (n0), we have adopted the method proposed by Martinez et al.50 We set the fluctuation, δ, at 20% of the average number of molecules (n0).
In Table 3, we have shown the results of our cluster structure analyses for all systems. It can be seen that the difference between the values of n − n0 is higher than the corresponding δ value for all systems considered here (except for system S5 at 285 K temperature) indicating that neopentane has some propensity to aggregating in pure water as well as in aqueous trehalose solutions. The much lower aggregation propensity of the neopentane molecules for system S5 at 285 K acts as a corroborative evidence of what we observed in the free energy difference plot (Fig. 3). For a fixed temperature, the addition of trehalose molecules causes a drop in the n − n0 value and the drop is much sharper as one moves from system S3 to S5 through system S4. Moreover, the difference in the value of n − n0 and δ decreases as the concentration increases. On the other hand, for a particular trehalose concentration, as the temperature increases there is an increase in the n − n0 value for all systems except for systems S0 and S1 in which the n − n0 values are essentially insensitive to a temperature change. These findings are consistent with our calculated association constant values for the different systems discussed above. Here, we mention that for concentrated trehalose solutions, a substantial drop in the number of trehalose molecules and a sharp rise in the number of water molecules in the neopentane solvation shell is observed at lower temperature. On the other hand, at higher temperature we observe much less exclusion of trehalose molecules from the neopentane surface and a smaller number of water molecules in the neopentane solvation shell. These findings are discussed below.
Table 3 Neopentane cluster size for the different systems. n0, n and δ are the average number of neopentane molecules, the number of neopentane molecules in the first coordination shell of a reference neopentane molecule and the fluctuation in n0, respectively
| System |
Temperature (K) |
n |
n0 |
δ |
n − n0 |
Remark |
| S0 |
285 K |
6.07 |
1.04 |
0.21 |
5.03 |
High |
| S0 |
300 K |
6.07 |
1.07 |
0.21 |
5.00 |
High |
| S0 |
315 K |
5.98 |
1.10 |
0.22 |
4.88 |
High |
| S0 |
330 K |
5.99 |
1.05 |
0.21 |
4.94 |
High |
| S0 |
345 K |
5.95 |
1.07 |
0.21 |
4.88 |
High |
| S1 |
285 K |
5.84 |
0.96 |
0.19 |
4.88 |
High |
| S1 |
300 K |
5.63 |
0.99 |
0.20 |
4.63 |
High |
| S1 |
315 K |
5.73 |
0.98 |
0.20 |
4.75 |
High |
| S1 |
330 K |
5.94 |
1.01 |
0.20 |
4.93 |
High |
| S1 |
345 K |
5.84 |
0.96 |
0.19 |
4.88 |
High |
| S2 |
285 K |
5.07 |
0.90 |
0.18 |
4.17 |
High |
| S2 |
300 K |
4.93 |
0.90 |
0.18 |
4.03 |
High |
| S2 |
315 K |
5.67 |
0.95 |
0.19 |
4.72 |
High |
| S2 |
330 K |
5.67 |
0.90 |
0.18 |
4.77 |
High |
| S2 |
345 K |
5.72 |
0.93 |
0.18 |
4.79 |
High |
| S3 |
285 K |
3.82 |
0.72 |
0.14 |
3.10 |
High |
| S3 |
300 K |
3.89 |
0.72 |
0.14 |
3.17 |
High |
| S3 |
315 K |
4.40 |
0.74 |
0.15 |
3.66 |
High |
| S3 |
330 K |
4.43 |
0.73 |
0.15 |
3.70 |
High |
| S3 |
345 K |
4.93 |
0.78 |
0.16 |
4.15 |
High |
| S4 |
285 K |
2.06 |
0.60 |
0.12 |
1.46 |
High |
| S4 |
300 K |
2.37 |
0.63 |
0.13 |
1.74 |
High |
| S4 |
315 K |
3.42 |
0.65 |
0.13 |
2.77 |
High |
| S4 |
330 K |
2.71 |
0.61 |
0.13 |
2.05 |
High |
| S4 |
345 K |
4.16 |
0.54 |
0.12 |
3.55 |
High |
| S5 |
285 K |
0.59 |
0.51 |
0.10 |
0.08 |
Low |
| S5 |
300 K |
1.16 |
0.52 |
0.10 |
0.64 |
High |
| S5 |
315 K |
1.18 |
0.58 |
0.12 |
0.60 |
High |
| S5 |
330 K |
1.51 |
0.55 |
0.11 |
0.96 |
High |
| S5 |
345 K |
1.99 |
0.54 |
0.11 |
1.45 |
High |
C. Site–site radial distribution function
In order to get an explicit picture of the effects of trehalose and the temperature change on the solvation of neopentane molecules, we have computed selected site–site radial distribution functions involving neopentane and different solution species (water and trehalose) and also between different solution species.
Fig. 4 displays the rdfs involving the neopentane central carbon atom and the oxygen atom of water (Ow). Focusing on the hydration of neopentane at 285 K in pure water (Fig. 4 (a)), first we observed that the neopentane–water rdf starts to rise at 3.35 Å and reaches a maximum at 4.85 Å. Hence, below 4.85 Å there is actually an exclusion of water molecules from the solvation shell of neopentane. The first minimum appears at 6.35 Å indicating the outer limit of the first hydration shell of neopentane. In pure water, we find that the water density is low in the first hydration shell of neopentane and is only 0.96 times the bulk water density. Further, it can be seen that the height of the first peak decreases monotonically, without affecting very much the position of the first minimum when the temperature increases from 285 to 345 K. This low water density around neopentane compares well with that of the already reported results.46 The effects of trehalose on the neopentane–water rdf at different temperatures are shown in Fig. 4 (b–f). Considering the influence of temperature on the hydration of neopentane in the presence of trehalose, we find that for all systems considered here, the first peak height decreases gradually in the neopentane–water rdfs with increasing temperature without changing the first peak position, and this effect is more prominent for systems S3, S4 and S5. Further, for a particular temperature, as the trehalose concentration is increased, we find a sharp increase in the first peak height of the neopentane–water rdf and this effect is less pronounced at high temperatures. These findings suggest the solvation of the neopentane molecules by water is reduced in a concentrated trehalose solution when the temperature is high.
 |
| | Fig. 4 Site–site distribution functions involving the neopentane central carbon atom and the water oxygen atom. | |
It is a well-known fact that the extent of hydrophobic solvation of a hydrophobic moiety is intimately related to the number of solvent molecules around it. So, to analyze the solvation of a hydrophobic solute further, we have calculated the first shell coordination numbers (CNs) of the water molecules around the solute neopentane molecule. The values of CNs for different systems can be calculated as51
| |
 | (3) |
where
ρβ and
rc represent the number density of species
β and the position of the first minimum in the corresponding distribution function, respectively.
The number of water and trehalose molecules that are present in the first coordination shell of neopentane are calculated using eqn (3) and the same are presented in Table 4 and 5, respectively. Note that, in order to calculate CNs for water, we have considered the rdfs involving the neopentane central carbon atom and the water oxygen atom and the same for trehalose—the neopentane central carbon and glycosidic oxygen (O1) rdfs are used. For a fixed temperature with increasing the trehalose concentration though the first peak height in the corresponding neopentane–water rdfs increases (see Fig. 4) from Table 4 it can be seen that the number of the first shell water molecules around neopentane do not follow any particular trend. We trace this anomaly as due to a decrease in the water number density as trehalose is added. Since the trehalose molecules were added without replacing water molecules, the box volume increases with increasing the trehalose concentration leading to a decrease in the water number density. To consider this reduced water number density effect, we have calculated the coordination numbers assuming that the only change with added trehalose molecules comes through the number density of water and these values are shown in parentheses in Table 4. From this table it is apparent that for a particular temperature, the difference between the calculated and “expected” coordination number is small for low trehalose concentrations and this difference increases with further addition of trehalose molecules. Moreover, for systems S4 and S5, the difference between these two coordination number values decreases (mainly due to a sharp decrease in the calculated coordination number value) as the temperature is increased. For example, for system S5, the difference between estimated and “expected” coordination number value is 8.57 at 285 K and the value for the same at 345 K is 6.35. From these observations it appears that the trehalose-induced enhancement of hydrophobic solvation (by water) for systems S5 and S4 which occurs at low temperature is reduced as the temperature increases. The data in Table 5 show that for all temperatures, the number of trehalose molecules in the neopentane's first solvation shell decreases on addition of trehalose, suggesting exclusion of the trehalose molecules from the neopentane solvation shell. In the same table, we also present the “expected” number of trehalose around the solute neopentane by considering the trehalose number densities for different systems. As can be seen from the difference between actual and “expected” trehalose coordination number values, there is a sharp decrease in its value for systems S4 and S5 as the temperature increases from 285 to 345 K and this effect is less prominent for other systems. Thus, for concentrated solutions, the exclusion of trehalose molecules from the neopentane first solvation shell is greater at lower temperature as compared to that at higher temperature.
Table 4 Number of water molecules in the first solvation shell of a neopentane molecule. For a fixed temperature, the numbers given in parentheses represent the first shell water molecules if the only change with added trehalose came through the water number density change
| System |
285 K |
300 K |
315 K |
330 K |
345 K |
| S0 |
16.51 |
15.46 |
15.36 |
14.54 |
14.27 |
| S1 |
13.57 (15.31) |
13.78 (14.41) |
13.69 (14.12) |
12.67 (13.50) |
13.10 (13.27) |
| S2 |
13.63 (14.33) |
15.14 (13.49) |
12.13 (13.31) |
12.43 (12.54) |
12.30 (12.45) |
| S3 |
15.05 (11.87) |
13.96 (11.20) |
12.18 (11.06) |
12.56 (10.46) |
10.37 (10.39) |
| S4 |
15.93 (10.53) |
15.57 (10.26) |
13.32 (9.72) |
13.78 (9.21) |
12.80 (9.08) |
| S5 |
17.92 (9.37) |
15.96 (8.76) |
15.72 (8.63) |
13.95 (8.19) |
14.44 (8.09) |
Table 5 Number of trehalose molecules in the first solvation shell of a neopentane molecule. For a fixed temperature, the numbers given in parentheses represent the first shell trehalose molecules if the only change with added trehalose came through the trehalose number density change
| System |
285 K |
300 K |
315 K |
330 K |
345 K |
| S1 |
1.19 |
0.87 |
0.83 |
0.87 |
0.81 |
| S2 |
0.63 (2.23) |
0.55 (1.63) |
0.76 (1.56) |
0.57 (1.74) |
0.68 (1.52) |
| S3 |
0.38 (4.61) |
0.49 (3.38) |
0.46 (3.25) |
0.42 (3.37) |
0.50 (3.18) |
| S4 |
0.47 (6.14) |
0.43 (4.65) |
0.42 (4.29) |
0.47 (4.45) |
0.31 (4.15) |
| S5 |
0.44 (7.28) |
0.46 (5.29) |
0.39 (5.07) |
0.43 (5.28) |
0.41 (4.94) |
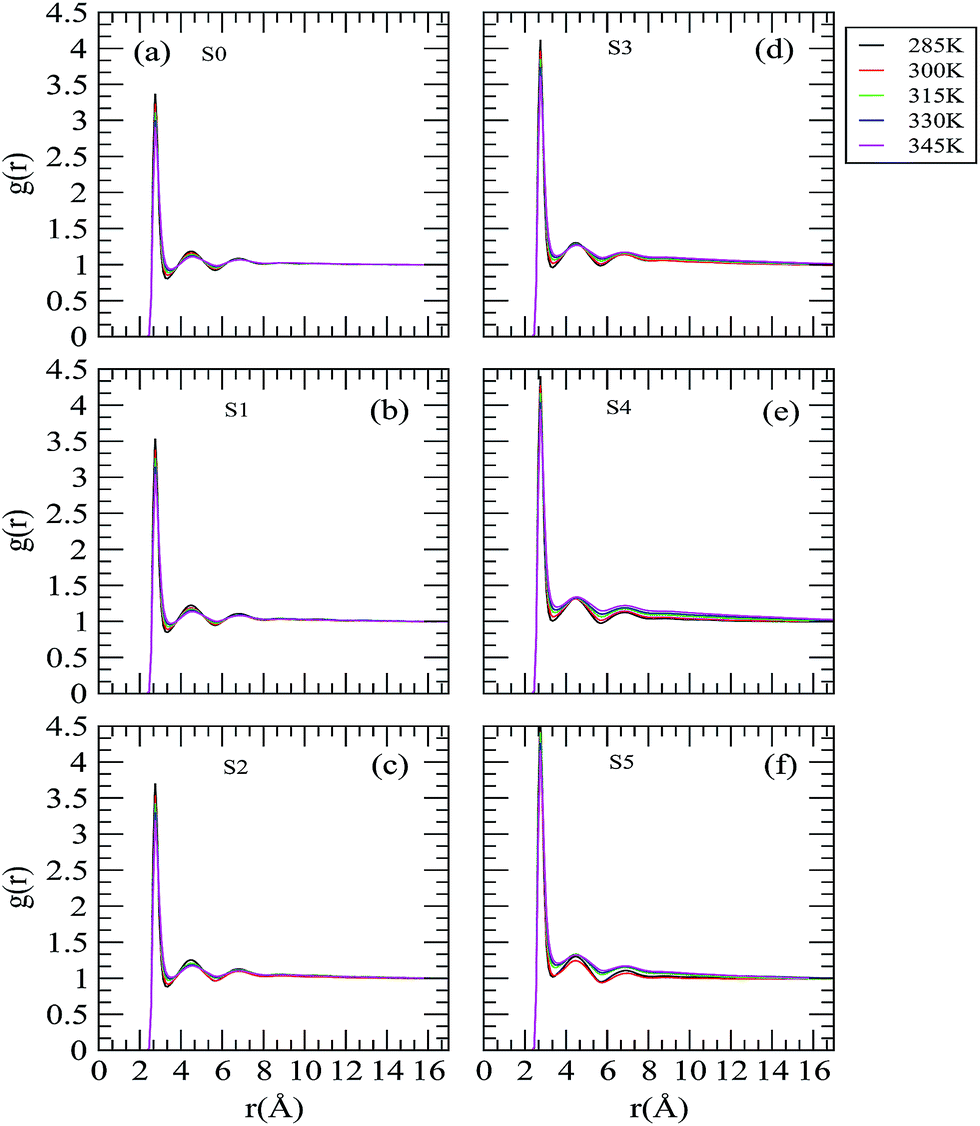
The influence of the trehalose molecules and the effect of the temperature change on the water structure, which indirectly provides the details of the hydrophobic solvation of neopentane molecules by water molecules, are reflected in the water oxygen–water oxygen (Ow–Ow) rdfs as shown in Fig. 5. The first and second peaks that appear at 2.75 and 4.45 Å characterize the H-bonded first neighbor and the tetrahedrally located second neighbor, respectively. The locations of these peaks in the Ow–Ow rdf are similar to those already reported in the literature.35,46,52,53 We find trehalose-induced enhancement in the first peak height of the Ow–Ow distribution function as we move from low to high trehalose concentration. Considering the effect of temperature at a fixed trehalose concentration, we observe that the locations of the first and second peak remain unaffected with increasing temperature. However, the height of the first and second peak decreases and the first valley becomes shallower as the temperature is increased. In this regard, we note that the temperature induced changes in the water–water correlation functions observed here are in accordance with the previous simulation studies of a water–methane system.54 These findings suggest that temperature and trehalose have two opposite effects with regard to changes in the water structure. Specifically, the water in the solutions becomes less structured as temperature is increased, which in turn makes the water molecules more mobile. On the other hand, addition of trehalose molecules makes the water molecules more structured and they become less mobile (see diffusion coefficient values discussed later).
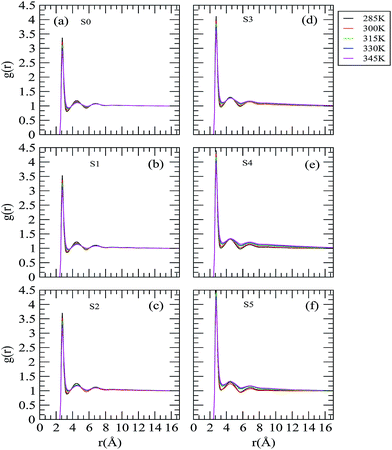 |
| | Fig. 5 Site–site distribution functions involving the oxygen atoms of water molecules. | |
An explicit picture of the interactions between water and trehalose molecules, which in turn influences the solvation of neopentane molecules, can be obtained by examining the selected site–site rdfs involving water and trehalose molecules. In Fig. 6, we show the typical rdfs involving one of the hydroxylic oxygens (O6) of trehalose and water oxygen. The distribution functions involving other hydroxylic oxygens of trehalose (O2, O3 and O4), glycosidic oxygen (O1) and acetalic ring oxygen (O5) atoms of trehalose with respect to water oxygen for different systems are also presented in Fig. 1–5 in the ESI† section. Note that the rdfs involving other hydroxylic trehalose oxygen atoms (O2, O3 and O4) and water oxygen (see Fig. 1–3 in the ESI† section) behave in a similar fashion as that of the O6–water oxygen rdf except for the fact that the second shell of the O4–water oxygen rdf is slightly less pronounced as compared to other hydroxylic oxygen–water oxygen distribution functions. In consistence with previously reported results,55–58 the observed characteristics of our calculated rdfs also suggest water mediated solvation of trehalose hydroxyl groups with well-defined nearest neighbor peaks in the corresponding trehalose O6 oxygen and water oxygen rdfs (see Fig. 6). The presence of a sharp peak in this rdf at 2.75 Å suggests that the water molecules are hydrogen bonded to the trehalose hydroxyl groups. On the other hand, the glycosidic oxygen (O1) atom (as well as O5 oxygen atom) of trehalose does not possess such a well defined first solvation peak (see Fig. 4 and 5 of ESI† section) implying that, due to a geometric constraint, the probability of finding water molecules in the vicinity of the glycosidic oxygen of (1-1)-linked disaccharide is very small.57,59 Considering first the effect of temperature at a fixed trehalose concentration we find that the first and second peak in the O6–water oxygen rdfs are essentially unchanged for systems S1 and S2 barring the fact that the first peak becomes slightly broadened as the temperature increases. On the other hand, for other systems, with increasing temperature, the height of both these peaks decreases and the position of the first peak remains essentially unaltered. Since the hydroxylic O6 atom can potentially form a hydrogen bond with water molecules, the above findings indicate that the temperature has little influence on the hydrogen bonding interactions between the water and trehalose molecules for systems S1 and S2 (discussed below). Further, the temperature dependent changes in the height and width of the first peak are reflected in the calculated number of the first shell water molecules around a trehalose molecule presented in Table 6. From this table it can be seen that for systems S1 and S2, the number of water molecules in the first solvation shell of trehalose increases as the temperature is increased, whereas it decreases for other systems. For a particular temperature, the addition of trehalose causes a small enhancement in the first peak height and a shallower first valley. Though we observe an increase in the first peak height of the rdfs, the number of the first shell water molecules around trehalose decrease as the trehalose concentration is increased. We attribute this is due to changes in the water number density as we move from system S1 to S5. The shallower first valley that appears at higher trehalose concentrations indicates breaking in the water–trehalose hydrogen bond network. This is further confirmed by our calculations of the average number of water–trehalose hydrogen bonds discussed below. We note, further, that at lower temperatures, the number of water molecules in the trehalose first solvation shell presented here matches well with the already reported values, considering the fact that we have used different temperatures and different trehalose concentrations55,60 and we employed a different method here in calculating the first shell coordination number when compared to other studies.61
 |
| | Fig. 6 Site–site radial distribution functions involving water oxygen and trehalose hydroxylic oxygen (O6). | |
Table 6 Number of the first shell water molecules around a trehalose molecule
| System |
285 K |
300 K |
315 K |
330 K |
345 K |
| S1 |
22.78 |
23.59 |
23.75 |
24.11 |
25.62 |
| S2 |
20.73 |
21.68 |
21.45 |
21.45 |
22.30 |
| S3 |
19.77 |
19.63 |
19.26 |
18.55 |
18.96 |
| S4 |
19.93 |
18.86 |
18.24 |
18.14 |
16.81 |
| S5 |
19.51 |
18.52 |
17.78 |
18.02 |
17.44 |
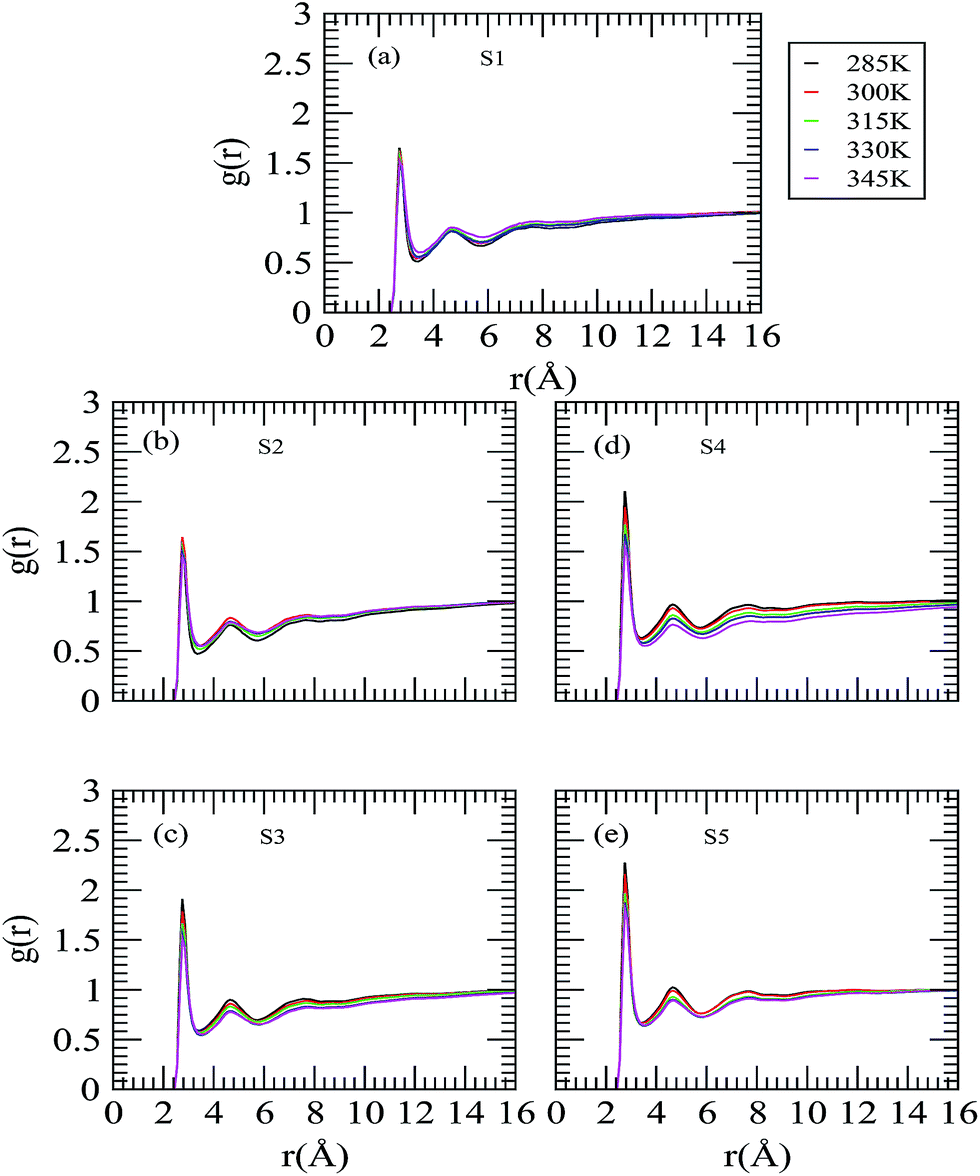
In an attempt to understand the role of trehalose molecules on neopentane aggregation, we have considered selected site–site rdfs of the central carbon of neopentane and trehalose O6 and O5 oxygen atoms with varying the trehalose concentration and temperature and the same are presented in Fig. 7 and 8. Keeping the temperature at a fixed value, considering the neopentane central atom-O6 rdf first (see Fig. 7) we observe: (a) depletion in the first peak height with increasing the trehalose concentration; (b) the second peak that was observed for system S1 starts disappearing on increasing the trehalose concentration. Examining the neopentane central atom-O5 rdf (see Fig. 8) we notice the disappearance of the first peak, which appeared at about 4.8 Å, and the appearance of a second peak at 7 Å as the trehalose concentration is increased. These results reveal that the propensity of the neopentane–trehalose interactions decreases as more and more trehalose is added. From these figures, it can also be seen that with increasing temperature the first peak height of these rdfs decreases substantially for systems S1 and S2 and for other systems there is essentially no strong qualitative temperature dependence. With regard to the effects of the trehalose concentration on the neopentane–O5 rdf, we observe a drop in the first peak height of these rdfs as trehalose is added, though except for systems S1 and S2 these functions are not strong functions of the trehalose concentrations. These results suggest exclusion of the trehalose molecules from the neopentane surface as the temperature and trehalose concentration increase. The weakening of the neopentane–trehalose interaction with the increasing temperature (and trehalose concentration) is further confirmed by our preferential interaction parameter (as discussed below) and is in agreement with the rise in the neopentane–water rdf as discussed above.
 |
| | Fig. 7 Radial distribution function involving the neopentane central carbon and the hydroxylic oxygen (O6) of trehalose. | |
 |
| | Fig. 8 Radial distribution function involving the neopentane central carbon and the acetalic ring oxygen (O5) of trehalose. | |
D. Preferential interaction
The above observation that increase in the trehalose concentration and rise in temperature cause exclusion of the trehalose molecules from the neopentane solvation shell and more and more water molecules are preferred by the neopentane molecule in its solvation shell has directed us to examine the difference between the local environment of a neopentane molecule to that of the bulk solution. This difference can be estimated by measuring the preferential binding parameter (τw/t(r)) as a function of the distance. Following previous works,62 τw/t(r) can be defined as:| | |
τw/t(r) = nw(r)/ntre(r) − Nw/Ntre
| (4) |
where nw(r) and ntre(r) are the number of water and trehalose molecules, respectively, at a distance r from the neopentane central carbon atom. N corresponds to the total number of molecules in the system with subscripts w and tre standing for water and trehalose molecules, respectively. So, τw/t(r), which represents the local ratio of the number of water to trehalose molecules minus their bulk ratio, is the measure of the binding parameter and it gives information on the deviation from an ideal solvation model. A negative value of τw/t indicates the preferential interaction of trehalose with the neopentane molecule over water and its positive value implies preferential hydration or preferential exclusion of trehalose in that region. Here we note that, though the transfer chemical potentials depend on the global thermodynamic preferential interaction parameters (as reported by the solvation experts Timasheff, Schellman and Parsegian63–66), in this work our goal is not to correlate our estimated τw/t(r) values with the transfer chemical potentials. Thus, our calculation of τw/t(r) gives only a qualitative idea of the preferential accumulation or exclusion of a solution species (over the second one) around a solute molecule at a distance r.
Fig. 9 shows the changes in τw/t for the neopentane central atom as a function of the distance. The initial positive numerical value of τw/t at a shorter distance is directly related to the relatively larger exclusion radius of the trehalose molecules. Considering system S1 at 285 K temperature first, we observe that the local ratio becomes equal to the bulk ratio at about 5.25 Å and then the τw/t value starts decreasing with the distance and reaches a minimum at 6.2 Å where it becomes negative. We further notice that, for this system, with increasing temperature τw/t increases slightly. On considering the effect of trehalose, it can be seen that as the trehalose concentration increases, the value of τw/t becomes more positive than system S1, suggesting exclusion of the trehalose molecules (and preference of the water molecules) from the neopentane surface. Moreover, for systems S3, S4 and S5 the value of τw/t is close to zero indicating both water and trehalose molecules are equally preferred by neopentane and the relative distribution of water to trehalose is more ideal-like. Further, for these three systems, the temperature has essentially no influence on the τw/t value.
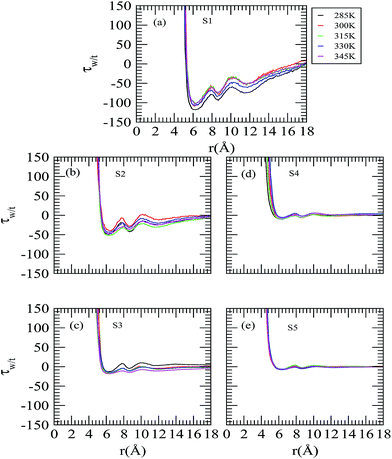 |
| | Fig. 9 Preferential interaction parameters around the neopentane molecule as a function of the temperature and concentration. | |
E. Hydrogen bonding
As observed in the water–trehalose rdfs (discussed above), due to the presence of hydroxyl groups, in aqueous solution a trehalose molecule possesses high affinity to form hydrogen bonds with water and also with other trehalose molecules. The water structure and the trehalose–trehalose interactions are greatly affected by the formation of hydrogen bonds between the water and trehalose molecules, and subsequently this may impact on the role of the trehalose molecule (as a protective co-solute) to biomacromolecules.67,68 The formation of a hydrogen bond is considered if the following conditions are satisfied simultaneously:
In brief, a pair of solution species is considered to be hydrogen bonded if the oxygen–oxygen distance is less than (or equal to) 3.4 Å while simultaneously the hydrogen–oxygen distance is less than (or equal to) 2.4 Å and the oxygen–hydrogen–oxygen angle is greater than (or equal to) 120°. The geometric criteria used above for defining the hydrogen bonds between water–water and water–trehalose are further verified by the probability contour plots as a function of the O–H⋯O angle and the H⋯O distance for system S5 at 300 K (see Fig. 6 in ESI†). From these plots, it can be seen that the hydrogen bond geometric criteria used in our study consider the region of high probability density for the formation of strong hydrogen bonds. For all systems, the average number of hydrogen bonds between trehalose–trehalose, water–water and water–trehalose (per trehalose) are shown in Table 7. We note that, for system S1 at 300 K temperature, the estimated number of water–trehalose hydrogen bonds per trehalose presented in this study match well the results by Donnamaria et al.56 On the other hand, by using the same hydrogen bonding criteria, Liu et al.69 reported that the average trehalose–water (per trehalose) hydrogen bond number is 22.4 for the TIP3P water model. This anomaly in the hydrogen bond number can be attributed to the use of a different water model in their study and to the presence of solute neopentane molecules and the higher trehalose concentration used in our study. Further, with increasing temperature, as expected, Guillot et al.70 observed a decrease in the average number of water–water hydrogen bonds. In the present study, our results (see Table 7) also show that the average number of water–water hydrogen bonds decreases monotonously for low trehalose concentrations when the temperature is increased. Surprisingly, when the trehalose concentration is very high (e.g., for systems S4 and S5), the average number of water–water hydrogen bonds is insensitive to a temperature change. Considering the water–trehalose (per trehalose) hydrogen bond number for low trehalose concentrations, we find that these hydrogen bonds are not a strong function of the temperature. On the other hand, for systems S4 and S5, the average number of water–trehalose (per trehalose) hydrogen bonds decreases with increasing the temperature. From these findings we make the following conclusions: (a) when the trehalose concentration is low, the temperature induced breaking of water–water hydrogen bonds enhances the hydrophobic solvation of a neopentane molecule (by water). (b) For higher trehalose concentrations, the temperature induced breaking of water–trehalose hydrogen bonds causes a release of some water and trehalose molecules. Some of these free water molecules form water–water hydrogen bonds and as a result the average number of water–water hydrogen remains constant that would otherwise decrease as the temperature is increased. These observations imply indirectly that for systems S4 and S5, the trehalose induced enhancement of the hydrophobic solvation of a neopentane molecule (by water) observed at low temperature, is reduced as the temperature increases. It, further, acts as a corroborative evidence of what we observe in the neopentane–water rdfs (discussed above). (c) For systems S4 and S5, some of the trehalose molecules that were released due to temperature induced breaking of the water–trehalose hydrogen bonds form hydrogen bonds with the like molecules. As a result, we observe enhancement in the trehalose–trehalose hydrogen bonds for these systems as the temperature is increased. With regard to the trehalose–trehalose hydrogen bonds, we find that for a fixed temperature, the addition of trehalose molecules increases the average number of trehalose–trehalose hydrogen bonds.
Table 7 Average number of water–water (per water), water–trehalose (per trehalose) and trehalose–trehalose (per trehalose) hydrogen bonds for different systems
| Systems |
285 K |
300 K |
315 K |
330 K |
345 K |
| HBwater–water |
| S0 |
3.51 |
3.48 |
3.45 |
3.40 |
3.36 |
| S1 |
3.43 |
3.38 |
3.35 |
3.32 |
3.28 |
| S2 |
3.33 |
3.31 |
3.26 |
3.26 |
3.20 |
| S3 |
3.07 |
3.07 |
3.11 |
3.04 |
2.98 |
| S4 |
2.86 |
2.83 |
2.85 |
2.84 |
2.89 |
| S5 |
2.61 |
2.68 |
2.67 |
2.67 |
2.68 |
| |
| HBwater–trehalose |
| S1 |
11.87 |
13.17 |
12.23 |
11.86 |
12.56 |
| S2 |
11.61 |
11.44 |
11.72 |
10.56 |
11.09 |
| S3 |
11.39 |
10.99 |
10.23 |
9.53 |
9.98 |
| S4 |
11.08 |
10.64 |
9.91 |
9.63 |
8.64 |
| S5 |
10.94 |
10.27 |
9.70 |
9.58 |
9.03 |
| |
| HBtrehalose–trehalose |
| S1 |
0.68 |
0.84 |
0.78 |
0.75 |
0.66 |
| S2 |
1.51 |
1.38 |
1.52 |
1.22 |
1.23 |
| S3 |
1.89 |
1.84 |
1.73 |
2.02 |
2.16 |
| S4 |
2.01 |
2.25 |
2.43 |
2.48 |
2.34 |
| S5 |
2.28 |
2.27 |
2.64 |
2.47 |
2.48 |
F. Trehalose clusters
As observed above, for a fixed temperature, the increase in the trehalose concentration causes enhancement in the average number of trehalose–trehalose hydrogen bonds. As a result, the association of trehalose molecules takes place.68,71 Moreover, Molinero et al.72 reported that the dynamical properties of the solution species are greatly affected due to formation of an extended sugar–sugar hydrogen bond network. Thus, in order to examine the effect of the trehalose concentration on its self-aggregation and the possible influence of the temperature on it, it is important to estimate the mean trehalose cluster size. We consider a trehalose cluster is an assembly of like molecules that are connected to each other with at least one hydrogen bond. The mean trehalose cluster size, (〈ntre〉), can be defined as follows:where ntre and Ptre are the number of trehalose molecules in a given cluster and its probability of formation, respectively. The normalized mean trehalose cluster sizes, 〈ntre〉/Ntre, (where Ntre is the number of trehalose molecules in a system) for systems S1 and S5 at different temperatures are shown in Fig. 10(a). Notwithstanding the error bars, it can be seen that the value of 〈ntre〉/Ntre for system S5 is much higher than that of system S1 for all temperatures, suggesting that on addition of trehalose, the average trehalose cluster size increases. Furthermore, for system S5 the value of 〈ntre〉/Ntre reaches 0.68 at 285 K, implying that even for the highest trehalose concentration (and lowest temperature) considered in this study, the percolation of the trehalose hydrogen bond network is yet to be achieved. In this context we note that Lerbret et al.71 reported the percolation of the trehalose hydrogen bond network (in a water–trehalose system) at 66% trehalose concentration. This anomaly is attributed to the use of a large number of neopentane molecules in our study. From this figure, we can further notice that the value of 〈ntre〉/Ntre for both systems decreases with increasing the temperature, indicating the formation of a greater number of smaller clusters from the breaking of the higher order trehalose cluster. In Fig. 10(b) and (c), considering C1–O1–C1 as a reference coordinate system, we show the projection of the spatial distribution function of the trehalose–trehalose interaction around a reference trehalose molecule for system S5 for the highest and lowest temperatures considered here. We observe that there is an overlap between the high probability density region for directly hydrogen bonded trehalose molecules and the high density region up to 10 Å and the effect is much more prominent for lower temperature when compared to higher temperature. This fact confirms the observations of the cluster structure analyses discussed above and further reveals the formation of hydrogen bond mediated trehalose clusters present in the system.
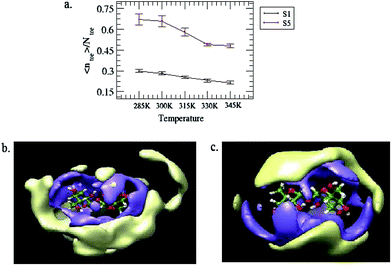 |
| | Fig. 10 (a) Normalized mean trehalose cluster size, 〈ntre〉/Ntre for systems S1 and S5 at different temperatures. The standard errors are calculated using a block average over 1 ns. Sections (b) and (c) represent a projection of the spatial distribution function of the trehalose–trehalose interaction for system S5 at temperatures 285 and 345 K, respectively. The isosurface in blue represents the higher probability density of direct hydrogen bonding interactions with the central reference trehalose molecule and the isosurface in yellow corresponds to a high density region of up to 10 Å, from the reference trehalose molecule. | |
G. Diffusion coefficients
It has already been observed that trehalose has a profound effect on the dynamical properties of different solution species present in an aqueous trehalose solution. This is due to the fact that the concentrated aqueous trehalose solution has a very high glass transition temperature and it forms a highly viscous glassy matrix which helps protect the biomolecule in its biological conformation like an insect trapped in amber.11 In view of this, we have estimated the translation diffusion coefficients of different solution species for different systems by using the widely used Einstein relation.| |
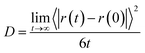 | (6) |
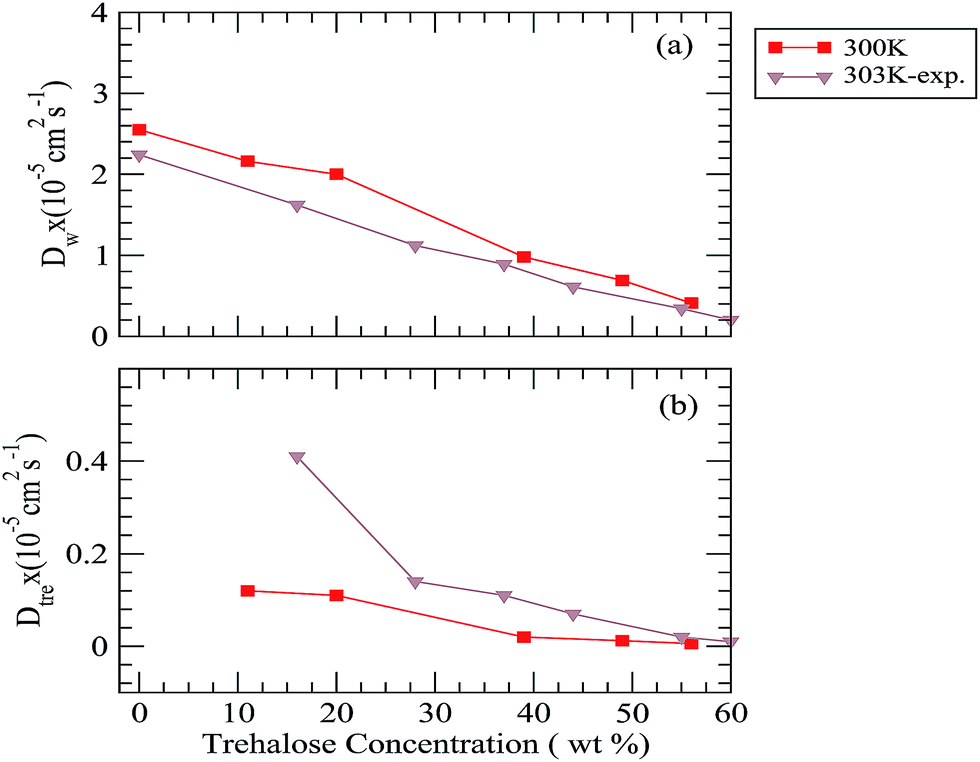
Using the above eqn (6), the diffusion coefficients (D) can be calculated from the long time slope of mean square displacement (MSD). The results obtained for water, trehalose together with the available experimental diffusion coefficient values73 at 300 K temperature are shown in Fig. 11 and the same for other temperatures are shown in Fig. 7 in the ESI.† The diffusion coefficient values of neopentane for different systems are also presented in Table I in the ESI.† For a fixed temperature the addition of trehalose reduces the diffusion coefficients of both water (Dw) and trehalose (Dtre). However, the drop in the diffusion coefficient values is more pronounced for trehalose. For example, at 285 K, Dw × 105 is reduced from 1.58 to 0.24 (approximately 6.5 times drop) as one moves from system S1 to S5, whereas for the trehalose molecules we observe a 20 times drop in the Dtre value (from 0.06 of system S1 to 0.003 for system S5). Moreover, in accordance with the previously reported results of NMR experiments,73 for all temperatures considered here, we also find a drop in the value of Dtre:Dw as the trehalose concentration increases. We note that the sharp drop in the Dtre values, particularly at higher trehalose concentrations, is in good agreement with the sharp increase in the viscosity values reported earlier.74 In this regard it is worth noting that the formation of stable water–trehalose hydrogen bonds and trehalose which induced a significant decrease in the diffusion coefficient of water molecules present near 5.5 Å of a solute molecule has already been reported elsewhere.75 Further, the results of a recent terahertz absorption measurement study76 also suggest trehalose-induced retardation of the translational and rotational motion of water molecules. With regard to the effect of temperature, we find that for a particular trehalose concentration, as expected, the values of both Dw and Dtre increase with increasing temperature. We note that our estimated diffusion coefficient values for water and trehalose compare reasonably well with the experimental findings. In this context we note that the results of the photo correlation spectroscopy and viscosity measurements study of an aqueous trehalose solution also show a similar trend of the diffusion coefficient change with respect to the temperature change.77–79 The diffusion coefficient of neopentane (Dnp) decreases with increasing the trehalose concentration and it increases as the temperature is increased, however, we note that the change in its value is not a strong function of either the trehalose concentration or the temperature. Furthermore, the non-appearance of the so-called “boson” peak in the log–log plot of the mean square displacement of trehalose (see Fig. 8 in ESI†) even for the highest trehalose concentration and lowest temperature considered here, suggests that the most concentrated trehalose solution in our study is still above the glass transition temperature.
 |
| | Fig. 11 Diffusion coefficient of (a) water and (b) trehalose for different systems. The experimental diffusion coefficient values are taken from ref. 73. | |
IV. Summary and conclusions
In this article, we discuss MD simulation results for a model neopentane–water–trehalose ternary solution. We considered six different trehalose concentrations and for each trehalose concentration five different temperature values were used. From the neopentane–neopentane potentials of the mean force we found trehalose induced destabilization of the neopentane contact pair at low temperature. Interestingly, concentrated trehalose solution helps stabilize the contact pair state of neopentane at high temperature. Thus, at high concentration the trehalose molecules act as a stabilizing co-solute when the temperature is also very high. These observations are further supported by the estimated association constant values and cluster structure analyses for different systems. To provide a molecular level understanding of the influence of trehalose and the temperature change, we have calculated selected site–site rdfs involving different solution species. In ambient conditions, the neopentane–water rdf showed a decreased structure in the more concentrated trehalose solutions, suggesting efficient hydration of neopentane by water. On the other hand, the first peak height of the neopentane–water rdf is reduced gradually on increasing the temperature and this effect is very prominent for concentrated trehalose solution. These observations imply that trehalose induced solvation of neopentane (by water) in concentrated solution that was observed at lower temperatures is getting reduced as the temperature is increased. This fact is further confirmed by calculating the number of water molecules present in the first solvation shell of neopentane. With regard to neopentane–trehalose interactions, we found that an addition of more trehalose molecules causes exclusion of trehalose molecules from the neopentane surface, supporting the preferential hydration hypothesis.16 In this context, it is worth noting that exclusion of trehalose from the protein domain has already been reported earlier.17
Investigation of the hydrogen bond properties reveals that with increasing the temperature though the average number of water–water hydrogen bonds decreases remarkably for systems with low trehalose concentrations, for concentrated trehalose solution the temperature has no influence on the average number of water–water hydrogen bonds. With regard to the water–trehalose hydrogen bonds we found that for concentrated trehalose solution, the average number of this type of hydrogen bonds decreases with increasing the temperature. By examining the effect of the trehalose concentration and temperature change on the trehalose clusters we observed breaking (or formation) of higher order trehalose clusters as the temperature (or trehalose concentration) is increased. The value of the normalized mean cluster size indicates that percolation of the trehalose hydrogen bond network has not been achieved even for the highest concentration and the lowest temperature considered in this study. The calculation of the diffusion coefficients of different solution species showed trehalose induced slowing down of the translational motion of all solution species and the effect is much more pronounced for trehalose than for water (or neopentane). For a fixed trehalose concentration, as expected, we observed an enhancement in the diffusion coefficient values as temperature increases. Consistent with the NMR experiment73 we also found that for a fixed temperature, the ratio of the diffusion coefficient values of water and trehalose increases with increasing the trehalose concentration. Further, our calculated log–log plot of the mean-squared displacement vs. time does not show an appearance of a boson peak, as reported previously, suggesting that the most concentrated trehalose solution considered in our study is still above the glass transition temperature.58
Acknowledgements
Financial support from the Board of Research in Nuclear Sciences (BRNS), Govt. of India, is gratefully acknowledged. Part of the work was enabled by using the C-DAC (Pune) computational facility.
References
- J. H. Crowe, L. M. Crowe, A. E. Oliver, N. Tsvetkova, W. Wolkers and F. Tablin, Cryobiology, 2001, 43, 89–105 CrossRef CAS PubMed.
- J. Darnell, H. Lodish and D. Baltimore, Molecular Cell Biology, Scientific American Books, New York, 2nd edn, 1990 Search PubMed.
- A. S. Sussmann and H. O. Halvorson, Spores: Their Dormancy and Germination, Harper and Row, New York, 1966 Search PubMed.
- F. Franks, Biophysics and Biochemistry at low Temperatures, Cambridge University Press, Cambridge, 1985 Search PubMed.
- J. H. Crowe, J. F. Carpenter and L. M. Crowe, Annu. Rev. Physiol., 1998, 60, 73–103 CrossRef CAS PubMed.
- J. F. Carpenter, M. J. Pikal, B. S. Chang and T. W. Randolph, Pharm. Res., 1997, 14, 969–975 CrossRef CAS.
- T. Higashiyama, Pure Appl. Chem., 2002, 74, 1263–1269 CrossRef CAS.
- A. B. Richards, S. Krakowka, L. B. Dexter, H. Schmid, A. P. M. Wolterbeek, D. H. Waalkens-Berendsen, A. Shigoyuki and M. Kurimoto, Food Chem. Toxicol., 2002, 40, 871–898 CrossRef CAS.
- J. H. Crowe, L. M. Crowe, W. F. Wolkers, A. E. Oliver, X. Ma, J.-H. Auh, M. Tang, S. Zhu, J. Norris and F. Tablin, Integr. Comp. Biol., 2005, 45, 810–820 CrossRef CAS PubMed.
- A. D. Elbein, Y. T. Pan, I. Pastuszak and D. Carroll, Glycobiology, 2003, 13, 17R–27R CrossRef CAS PubMed.
- J. L. Green and C. A. Angell, J. Phys. Chem., 1989, 93, 2880–2882 CrossRef CAS.
- C. A. Angell, Chem. Rev., 2002, 102, 2627–2650 CrossRef CAS PubMed.
- J. F. Carpenter and J. H. Crowe, Biochemistry, 1989, 28, 3916–3922 CrossRef CAS.
- J. H. Crowe, S. B. Leslie and L. M. Crowe, Cryobiology, 1994, 31, 355–366 CrossRef CAS PubMed.
- S. D. Allison, B. Chang, T. W. Randolph and J. F. Carpenter, Arch. Biochem. Biophys., 1999, 365, 289–298 CrossRef CAS PubMed.
- P. S. Belton and A. M. Gil, Biopolymers, 1994, 34, 957–961 CrossRef CAS PubMed.
- S. N. Timasheff, Biochemistry, 2002, 41, 13473–13482 CrossRef CAS.
- S. Paul and S. Paul, J. Chem. Phys., 2013, 139, 044508-1–044508-9 CrossRef PubMed.
- G. Cottone, G. Ciccotti and L. Cordone, J. Chem. Phys., 2002, 117, 9862–9866 CrossRef CAS PubMed.
- C. S. Pereira and P. H. Hünenberger, J. Phys. Chem. B, 2006, 110, 15572–15581 CrossRef CAS PubMed.
- A. S. Reddy, A. Izmitli and J. J. de Pablo, J. Chem. Phys., 2009, 131, 085101-1–085101-8 CrossRef PubMed.
- R. D. Lins, C. S. Pereira and P. H. Hünenberger, Proteins: Struct., Funct., Bioinf., 2004, 55, 177–186 CrossRef CAS PubMed.
- S. J. Prestrelski, N. Tedeschi, T. Arakawa and J. F. Carpenter, Biophys. J., 1993, 65, 661–671 CrossRef CAS.
- L. Cordone, M. Ferrand, E. Vitrano and G. Zaccai, Biophys. J., 1999, 76, 1043–1047 CrossRef CAS.
- J. K. Kaushik and R. Bhat, J. Biol. Chem., 2003, 278, 26458–26465 CrossRef CAS PubMed.
- C. Narayanan and C. L. Dias, J. Chem. Phys., 2013, 139, 115103-1–115103-7 CrossRef PubMed.
- W. Wang, Int. J. Pharm., 1999, 185, 129–188 CrossRef CAS.
- R. A. Dimitrov and R. R. Crichton, Proteins: Struct., Funct., Bioinf., 1997, 27, 576–596 CrossRef CAS.
- W. Kauzmann, Adv. Protein Chem., 1959, 14, 1–63 CrossRef CAS.
- K. A. Dill, Biochemistry, 1990, 29, 7133–7155 CrossRef CAS.
- H. Li, C. Tang and N. S. Wingreen, Phys. Rev. Lett., 1997, 79, 765–768 CrossRef CAS.
- K. N. Kirschner, A. B. Yongye, S. M. Tschample, J. Gonzaa'lez-Outeiriňo, C. R. Daniels, B. L. Foley and R. J. Woods, J. Comput. Chem., 2008, 29, 622–655 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, J. Phys. Chem., 1987, 91, 6269–6271 CrossRef CAS.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- R. Sarma and S. Paul, J. Chem. Phys., 2011, 135, 174501-1–174501-12 CrossRef PubMed.
- X. Huang, C. J. Margulis and B. J. Berne, J. Phys. Chem. B, 2003, 107, 11742–11748 CrossRef CAS.
- D. A. Case, T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, M. Crowley, R. C. Walker, W. Zhang, K. M. Metz, B. Wang, S. Hayik, A. Roitberg, G. Seabra, I. Kolossvea'ry, K. F. Wong, F. Paesani, J. Vanicek, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, L. Yang, C. Tan, J. Mongan, V. Hornak, G. Cui, D. H. Mathews, M. G. Seetin, C. Sagui, V. Babin, P. A. Kollman, AMBER 10, University of California, San Francisco, 2008 Search PubMed.
- C. A. Stortz, P. G. Jhonson, A. D. French and G. I. Csonka, Carbohydr. Res., 2009, 344, 2217–2228 CrossRef CAS PubMed.
- F. Corzana, M. S. Motawia, C. H. Du Oenhoat, S. Perez, S. M. Tschampel, R. J. Woods and S. B. Engelsen, J. Comput. Chem., 2004, 25, 573–586 CrossRef CAS PubMed.
- P. Mark and L. Nilsson, J. Phys. Chem. A, 2001, 105, 9954–9960 CrossRef CAS.
- V. A. Verde and R. K. Campen, J. Phys. Chem. B, 2011, 115, 7069–7084 CrossRef PubMed.
- L. Marti'nez, R. Andrade, E. G. Birgin and J. M. Marti'nez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684-1–3684-7 CrossRef PubMed.
- J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- R. Sarma and S. Paul, J. Chem. Phys., 2012, 136, 114510-1–114510-10 CrossRef PubMed.
- M.-E. Lee and N. F. A. v. d. Vegt, J. Am. Chem. Soc., 2006, 128, 4948–4949 CrossRef CAS PubMed.
- R. Sarma and S. Paul, J. Phys. Chem. B, 2012, 116, 2831–2841 CrossRef CAS PubMed.
- S. Lüdemann, H. Schreiber, R. Abseher and O. Steinhauser, J. Chem. Phys., 1996, 104, 286–295 CrossRef PubMed.
- H. L. Martinez, R. Ravi and S. C. Tucker, J. Chem. Phys., 1996, 104, 1067–1080 CrossRef CAS PubMed.
- S. E. Pagnotta, M. A. Ricci, F. Bruni, S. McLain and S. Magazù, Chem. Phys., 2008, 345, 159–163 CrossRef CAS PubMed.
- S. Chowdhuri and A. Chandra, J. Chem. Phys., 2001, 115, 3732–3741 CrossRef CAS PubMed.
- H. Wei, Y. Fan and Y. Q. Gao, J. Phys. Chem. B, 2010, 114, 557–568 CrossRef CAS PubMed.
- N. T. Skipper, Chem. Phys. Lett., 1993, 207, 424–429 CrossRef CAS.
- G. Bonanno, R. Noto and S. L. Fornili, J. Chem. Soc., Faraday Trans., 1998, 94, 2755–2762 RSC.
- M. C. Donnamaria, E. I. Howard and J. R. Grigera, J. Chem. Soc., Faraday Trans., 1994, 90, 2731–2735 RSC.
- M. Sakurai, M. Murata, Y. Inoue, A. Hino and S. Kobayashi, Bull. Chem. Soc. Jpn., 1997, 70, 847–858 CrossRef CAS.
- P. B. Conrad and J. J. de Pablo, J. Phys. Chem. A, 1999, 103, 4049–4055 CrossRef CAS.
- Y. Choi, K. W. Cho, K. Jeong and S. Jung, Carbohydr. Res., 2006, 341, 1020–1028 CrossRef CAS PubMed.
- S. Magazù, P. Migliardo, A. M. Musolino and M. T. Sciortino, J. Phys. Chem. B, 1997, 101, 2348–2351 CrossRef.
- S. B. Engelsen and S. Pérez, J. Phys. Chem. B, 2000, 104, 9301–9311 CrossRef CAS.
- R. Sarma and S. Paul, J. Phys. Chem. B, 2013, 117, 677–689 CrossRef CAS PubMed.
- H. Inoue and S. N. Timasheff, Biopolymers, 1972, 11, 737–743 CrossRef CAS PubMed.
- S. N. Timasheff, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9721–9726 CrossRef CAS PubMed.
- J. A. Schellman, Annu. Rev. Biophys. Biophys. Chem., 1987, 16, 115–137 CrossRef CAS PubMed.
- V. A. Parsegian, R. P. Rand and D. C. Rau, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3987–3992 CrossRef CAS.
- R. Giangiacomo, Food Chem., 2006, 96, 371–379 CrossRef CAS PubMed.
- L. Sapir and D. Harries, J. Phys. Chem. B, 2011, 115, 624–634 CrossRef CAS PubMed.
- Q. Liu, R. K. Schmidt, B. Teo, P. A. Karplus and J. W. Brady, J. Am. Chem. Soc., 1997, 119, 7851–7862 CrossRef CAS.
- B. Guillot and Y. Guissani, J. Chem. Phys., 1993, 99, 8075–8094 CrossRef CAS PubMed.
- A. Lerbret, P. Bordat, F. Affouard, M. Descamps and F. Migliardo, J. Phys. Chem. B, 2005, 109, 11046–11057 CrossRef CAS PubMed.
- V. Molinero, T. Çağin and W. A. Goddard III, Chem. Phys. Lett., 2003, 377, 469–474 CrossRef CAS.
- N. Ekdawi-sever, J. J. de Pablo, E. Feick and E. von Meerwall, J. Phys. Chem. A, 2003, 107, 936–943 CrossRef CAS.
- D. P. Miller, J. J. de Pablo and H. Corti, Pharm. Res., 1997, 14, 578–590 CrossRef CAS.
- S. L. Lee, P. G. Debenedetti and J. R. Errington, J. Chem. Phys., 2005, 122, 204511-1–204511-10 Search PubMed.
- M. Heyden, E. Bründermann, U. Heugen, G. Niehues, D. M. Leitner and M. Havenith, J. Am. Chem. Soc., 2008, 130, 5773–5779 CrossRef CAS PubMed.
- S. Magazu, G. Maisano, H. D. Middendorf, P. Migliardo, A. M. Musolino and V. Villari, J. Phys. Chem. B, 1998, 102, 2060–2063 CrossRef CAS.
- E. Iannilli, E. Tettamanti, L. Galantini and S. Magazù, J. Phys. Chem. B, 2001, 105, 12143–12149 CrossRef CAS.
- M. Rampp, C. Buttersack and H. D. Lüdemann, Carbohydr. Res., 2000, 328, 561–572 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Figures showing: (i) O2–water oxygen rdf, (ii) O3–water oxygen rdf, (iii) O4–water oxygen rdf, (iv) O1–water oxygen rdf, (v) O5–water oxygen rdf, (vi) probability distribution contour plots as a function of hydrogen bond angle (D-H-A) and acceptor-hydrogen (A-H) distance for system S5 at 300 K, (vii) diffusion coefficients of water and trehalose and (ii) log–log plot of mean-squared displacement (MSD) vs. time for (a) water and (b) trehalose for system S5 at 285 K temperature. Table showing: (i) diffusion coefficients of neopentane for different systems. See DOI: 10.1039/c4ra03678f |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.