
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, characterization and gas permeation properties of anthracene maleimide-based polymers of intrinsic microporosity†
Muntazim Munir Khana,
Gisela Bengtsona,
Silvio Neumanna,
Md. Mushfequr Rahmana,
Volker Abetzab and
Volkan Filiz*a
aHelmholtz-Zentrum Geesthacht, Institute of Polymer Research, Max-Planck-Str. 1, 21502 Geesthacht, Germany. E-mail: volkan.filiz@hzg.demailto
bUniversity of Hamburg, Institute of Physical Chemistry, Grindelallee 117, 20146 Hamburg, Germany
First published on 1st August 2014
Abstract
A series of new monomers containing dialkyl anthracene maleimide derivatives [4a,b(I–V)], which can be used as a precursor of a polymer of intrinsic microporosity (PIM) has been synthesized and characterized successfully. The homopolymers prepared via polycondensation with 2,3,5,6,-tetrafluoroterephthalonitrile (TFTPN) and their copolymers in combination with 5,5′,6,6′-tetrahydroxy-3,3,3′,3′-tetramethyl-1,1′-spirobisindane (TTSBI) were characterized by SEC, FT-IR, TGA, 1H-NMR, BET-surface area and gas transport properties. Compared to polymers derived from 4a(I–V) monomers, the homopolymers and copolymers obtained from 4b(I–V) show improved solubility in common organic solvents and have high average molecular weight. Therefore they are able to form robust and transparent films. The gas transport properties of homopolymers and copolymers of 4b(I–V) show enhanced selectivity compared to PIM-1 for gas pairs such as O2/N2, CO2/N2 and CO2/CH4, followed by a slight decrease in permeability. The introduction of anthracene maleimide units (especially 4bIII) in the copolymer leads to more efficient chain packing and gives the copolymer a similar pore width distribution as PIM-1. As a consequence, the introduction of anthracene maleimide enhanced the CO2 selectivity of copolymers, compared to previously reported film forming polymers. Therefore, these polymers might be useful for gas separations relying on CO2 selectivity.
1. Introduction
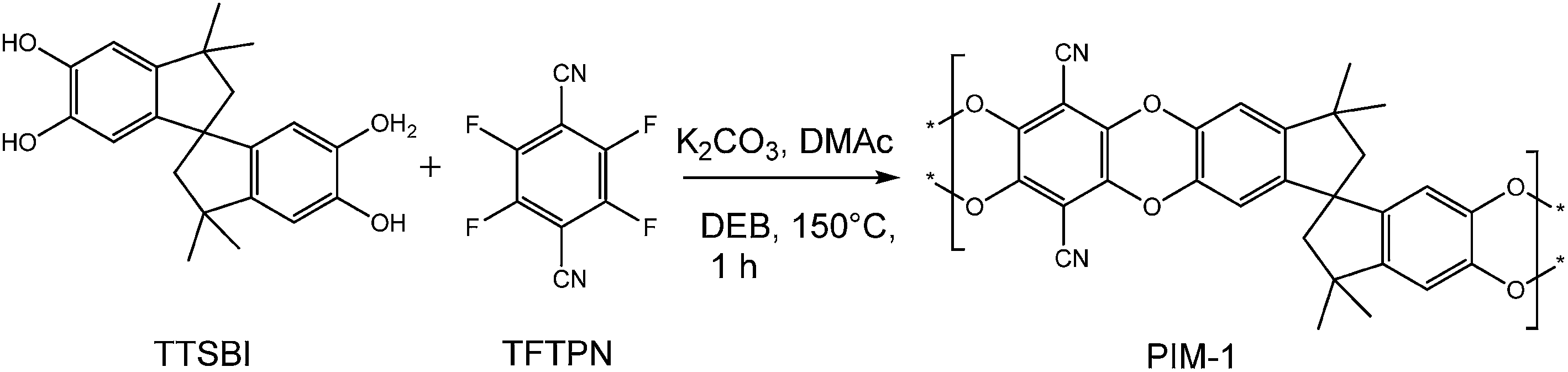
During the past years, microporous organic materials gained increasing interest as they have a high potential in a variety of applications like gas separation or storage. These microporous polymer-based materials can be obtained by the extensive crosslinking of solvent swollen polymers (e.g., hypercrosslinked polystyrenes)1 or by preparing polymer networks using rigid monomers.2–4 In addition, certain highly rigid, non-crosslinked polymers, for example poly(trimethylsilylpropyne),5,6 certain polyimides,7 and a number of fluorinated polymers,8 form solids with very large free volumes so that they behaving similarly to microporous materials. However, only a very limited number of microporous and soluble polymers are known so far.The most extensively studied purely organic materials with network and non-network structure, introduced by Budd et al., are termed as polymers of intrinsic microporosity (PIM), possessing good solubility in organic solvents, high thermal stability and high surface area. The intrinsic microporosity is caused by a rigid and contorted molecular structure which cannot fill space efficiently, leaving molecular sized-interconnected voids.9,10 This rigidity and contortion arises from the incorporation of non-linear ‘sites of contortion’ such as spirocyclic or triptycene units.11,12 Even compared with traditional widely used industrially microporous materials, such as zeolites or activated carbons. PIMs present a unique advantage, due to the combination of processability with a high degree of microporosity, which are attractive for applications such as adsorbents, gas separation and catalysis. Generally, PIMs can be easily prepared by polycondensation reaction (double aromatic nucleophilic substitution) using a combination of a tetrafunctional hydroxylated aromatic monomer and an activated tetrafluorinated or chlorinated aromatic monomer to form rigid dibenzodioxane linking groups resulting in ladder architecture. The first and most popular polymer of this type is referred to as PIM-1, derived by step-growth polymerization of 5,5′,6,6′-tetrahydroxy-3,3,3′,3′-tetra-methylspirobisindane (TTSBI) and 2,3,5,6,-tetrafluoroterephthalonitrile (TFTPN) (Scheme 1). Despite the tremendous progress in the field of PIMs synthesis, there is still a need for the development of these materials that can exhibit higher (and more stable) free volume and which can be tailored for other applications such as gas separation, gas storage and liquid filtration.13 This can be established via designing new monomers that will be utilized for polymer synthesis.
 | ||
| Scheme 1 Synthesis of PIM-1. | ||
One of the structural components used previously for making PIM is 9,10-dimethyl-9,10-ethano-9,10-dihydro-2,3,6,7-tetrahydroxy-anthracene (CO1).14,15 The resulting network polymer from the reaction between CO1 and TFTPN demonstrated an apparent BET surface area of 750 m2 g−1 with the microporosity presumably arising from the roof shaped of the CO1 units.14 Due to the potential for providing pre-formed concave binding sites of a fixed size and shape for specific adsorption, we are interested in synthesizing new examples of PIM with similar roof shaped components. Hence our attention was drawn to the anthracene maleimide unit, which has been exploited widely as a component of functional molecular systems including shape memory properties,16 chemical and biological activities,17–19 click reactions20,21 and chiral auxiliaries.22 To our knowledge, there is no literature available so far on anthracene maleimide based polymers. In the context of PIM, it was anticipated that the roof shaped structure of the anthracene maleimide unit should ensure greater polymer rigidity as compared to CO1 and different substituents on the maleic anhydride ring could frustrate further the polymer chain packing which might enhance the intrinsic microporosity. In the present work, we describe synthesis, characterization and gas transport properties of new polymers of intrinsic microporosity (PIM) derived from dialkyl anthracene maleimide monomers, polycondensated with 2,3,5,6,-tetrafluoroterephthalo-nitrile (TFTPN) as well as their copolymers in combination with tetrahydroxy monomer of PIM-1.
2. Experimental
2.1. Materials and methods
Most commercially available reagents were obtained from Sigma-Aldrich (Germany) and were used without further treatment. The exceptions: 5,5′,6,6′-tetrahydroxy-3,3,3′,3′-tetramethyl-1,1′-spirobisindane (TTSBI, 97%) was obtained from ABCR (Germany), and 2,3,5,6,-tetrafluoro-terephthalonitrile (TFTPN, 99%) was kindly donated by Lanxess (Germany). TFTPN was sublimated twice in vacuum prior to use. Potassium carbonate (K2CO3, > 99.5%) was dried overnight under vacuum at 120 °C and milled in a ball mill for 15 min. (Co)monomers 4a/b-I to V were prepared in our laboratory following the procedures described below. All reactions using air/moisture sensitive reagents were performed under argon atmosphere. NMR spectra were recorded on a Bruker AV300 NMR spectrometer operating at a field of 7 Tesla (300 MHz) using a 5 mm 1H/13C TXI probe and a sample temperature of 298 K. 1H spectra were recorded applying a 10 ms 90° pulse. 13C spectra were recorded using dept-45 and dept-q135 sequences employing a waltz-16 decoupling scheme. The relaxation delay was chosen so that the sample was fully relaxed. FT-Infrared spectra were recorded on attenuated total reflectance (ATR-diamond crystal) mode with a Bruker ALPHA FT-IR spectrometer. The transmission measurements were done in a spectral range of 400–4000 cm−1 with a resolution of 4 cm−1 and average of 64 scans. Thermogravimetric analysis (TGA) onset temperature measurements were obtained using a Netzsch TG209 F1 Iris instrument under argon flow (20 ml min−1) from 25 °C to 900 °C at 10 K min−1. The retro Diels–Alder reaction temperature was investigated using thermogravimetric analysis coupled with FT-IR. Molar mass distributions were determined by Size Exclusion Chromatography (SEC) with chloroform as solvent using polystyrene calibrated standards. Low temperature (77 K) nitrogen (N2) adsorption/desorption study of the polymer powder was undertaken using Rubotherm IsoSORP® instrument (Rubotherm GmbH, Bochum, Germany). Apparent surface area of the polymer was calculated from N2 adsorption data by multipoint BET analysis.2.2. Monomers synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.7) was added dropwise at 0–5 °C to a 3-fold volume H2SO4 (70%) and stirred for 20 min. The highly viscous lilac mixture was kept at room temperature for three days. The almost black mass was shovelled into ethanol–water (1:1), the resulting precipitate collected by filtration and dried in air. Two additional cleaning steps in chloroform and acetone give a light grey powder (50–60%). Mp: 325 °C. 1H-NMR (300 MHz, DMSO-d6): δ (ppm): 2.95 (s, 6H), 4.08 (s, 12H), 7.40 (s, 4H). FT-IR: 3019, 2996, 2963, 2833, 1633, 1495 (s), 1468, 1445, 1239, 1201, 1189, 1164 (s), 1013, 892, 828, 750 cm−1.
:1.7) was added dropwise at 0–5 °C to a 3-fold volume H2SO4 (70%) and stirred for 20 min. The highly viscous lilac mixture was kept at room temperature for three days. The almost black mass was shovelled into ethanol–water (1:1), the resulting precipitate collected by filtration and dried in air. Two additional cleaning steps in chloroform and acetone give a light grey powder (50–60%). Mp: 325 °C. 1H-NMR (300 MHz, DMSO-d6): δ (ppm): 2.95 (s, 6H), 4.08 (s, 12H), 7.40 (s, 4H). FT-IR: 3019, 2996, 2963, 2833, 1633, 1495 (s), 1468, 1445, 1239, 1201, 1189, 1164 (s), 1013, 892, 828, 750 cm−1.General procedure for demethylation. 3a-I (3.34 g, 6.68 mmol) was dissolved in 20 ml dichloromethane (DCM) and added slowly with cooling in an ice-bath to boron tribromide (1.26 ml, 13.37 mmol), dissolved in 40 ml DCM under argon atmosphere. The reaction mixture was stirred at room temperature for 18 h and then poured to water (1 litre). The acidic pH (=5) was neutralized with 1% NaOH till pH = 7–8. The precipitate was filtered and washed with copious amount of water, dried in vacuum at 60 °C to give a grey solid (2.68 g, 90.7%), Mp: >300 °C. 1H NMR (300 MHz, DMSO-d6): δ (ppm): 2.00 (s, 6H), 2.97 (s, 2H), 6.52 (dd, 2H), 6.55 (s, 2H), 6.65 (s, 2H), 7.33 (m, 3H), 8.73 (s, 4H), 13C NMR (75 MHz, DMSO-d6): δ (ppm): 16.1, 42.6, 53.0, 110.1, 110.4, 127.1, 128.7, 129.1, 132.5, 133.3, 136.7, 143.0, 143.3, 175.7. FT-IR: 3348, 2910, 1768, 1691, 1594, 1493, 1445, 1392, 1310 (s), 1236, 1210, 1184, 1153, 998, 757, 709, 691, 622 cm−1. C26H21NO6 calculated: C = 70.41%, H = 4.78%, N = 3.16%, O = 21.66%, found: C = 68.37%, H = 5.02%, N = 3.15%, O = 22.86%. TGA(onset) = 312 °C.
2.3. Polymer synthesis
Polymerization procedure. TTSBI, TFTPN and (or) comonomer were dissolved in DMAc to form a homogeneous solution in a three necked flask equipped with magnetic stirrer and argon inlet. Addition of K2CO3 (in excess of 1
:1.2 based on OH-component(s)) caused a solution colour change from yellow to orange. The reaction mixture was immersed in an oil bath preheated at 150 °C and within minutes the reaction mixture became increasingly viscous, then diethyl benzene (DEB) was added dropwise to maintain stirring and stirring was continued for 30 min. The hot reaction mixture was poured into methanol and the precipitated yellow solid was collected by filtration. The crude polymer was refluxed for 1 h in deionized water, filtrated and dried under vacuum at 120 °C for 48 h. The polymer was dissolved in chloroform and reprecipitated in methanol. In case the polymer product was insoluble in chloroform and dichlorobenzene, it was purified by washing with acetone.
2.4. Membrane preparation
Thick polymer films were prepared by solution casting method from polymer solution (concentration 1–2%, w/w) into accurately levelled Teflon circular dish. For low boiling solvent such as chloroform, the dish was covered by a lid and the system was gently purged using a stream of N2. After solvent evaporation, the prepared films were delaminated and conditioned by soaking in methanol for approximately 4 h. The methanol treated membranes were dried in high vacuum for 16 h at 120 °C. The thickness of the membranes was measured by a digital micrometer (Deltascopes MP2C) to 50–75 μm. Moreover, thin film composite (TFC) membranes for long term study were prepared on polyacrylonitrile (PAN) microporous support (average pore size of 25 nm and with 15% surface porosity) from polymer solution (conc. 0.5 or 1% w/w) in chloroform by an in house manufactured casting machine.2.5. Gas permeation measurement
Permeabilities of four pure gases (H2, N2, CO2, and CH4) were measured by a pressure increase time-lag apparatus at 30 °C. Permeability (P), diffusivity (D), solubility (S) and selectivity (α) for gases A and B were determined under steady state conditions by the following Equation:
 | (1) |
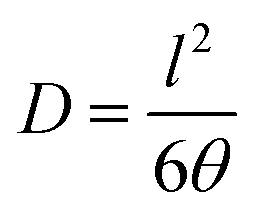 | (2) |
 | (3) |
Vp is the constant permeate volume, R is the gas constant, l is the film thickness, A is the effective area of membrane, Δt is the time for permeate pressure increase from pp1 to pp2, pf is the feed pressure, and θ is the time-lag. The solution–diffusion transport model26–29 was applied for discussing the gas transport properties of dense polymer membranes, and the selectivities of membranes for gas “A” relative to another gas “B” is the ratio of their permeabilities obtained from eqn (3).
3. Results and discussions
3.1. Monomer synthesis
The comonomers [4a(I–V), 4b(I–V)] were synthesized using a multistep synthetic route as shown in Scheme 2. The key intermediates for the synthesis of 4a-(I–V), 4b(I–V) are 1(a–b) and were prepared by slight modification to a published procedure,30,31 from the condensation reaction between the appropriate aldehyde (R) and 1,2-dimethoxybenzene in conc. H2SO4. The advantage of starting with methoxy groups is the enhanced solubility compared to hydroxyl groups and the high stability towards oxidation. The anthracene maleic anhydride derivatives 2(a–b) were prepared in high yield by Diels–Alder reaction between 1(a–b) and maleic anhydride in toluene using the procedure described by Milton C. Kloetzel.32 | ||
| Scheme 2 Synthetic route of monomer, reagents and conditions (A) when R: a = –CH3; conc. H2SO4, 18 h, room temperature; when R: b = –C4H9; acetonitrile, conc. H2SO4, room temperature, 2 h. (B) Maleic anhydride, toluene, 150 °C, 2 h. (C) Acetic anhydride, acetic acid, 130 °C, 24 h (D) BBr3, DCM, room temperature, 18 h. | ||
The imides at the maleic anhydride substituent [3a(I–V), 3b(I–V)] were prepared by reacting 2(a–b) with different aniline substituents (X) in the presence of glacial acetic acid and acetic anhydride for water removal to give the precursors [3a(I–V), 3b(I–V)]. The original synthesis of the maleimides was described by Leslie R.D. et al.33 Here it is slightly modified so as to allow for the simple introduction of substituted anilines and to provide symmetrical structure. Finally, the tetramethoxy precursors [3a(I–V), 3b(I–V)] were readily demethylated by BBr3 in CH2Cl2 to give the target tetrahydroxy monomers [4a-(I–V), 4b(I–V)]. The structures of the monomers were confirmed by FTIR, 1H NMR, 13C NMR, and elemental analysis.
1H NMR spectra of tetrahydroxy monomers 4a-I and 4b-I are shown in Fig. 1. 1H NMR signals corresponding to characteristic aromatic protons of monomer 4a-I are observed at 6.5, 6.6, 6.7 and 7.3 ppm, whereas aliphatic and maleic anhydride protons of 4a-I are observed at 2 and 2.9 ppm, respectively, as shown in Fig. 1i (4a-I). Monomer 4b-I shows the 1H NMR signals for aromatic protons at the same position as 4a-I due to identical framework. The aliphatic protons of the butyl groups in 4b-I range from 1.6 to 2.6 ppm, the characteristic protons originating from the former maleic anhydride shifted down field to 3.3 ppm because of the electron rich butyl groups, as depicted in Fig. 1ii (4b-I).
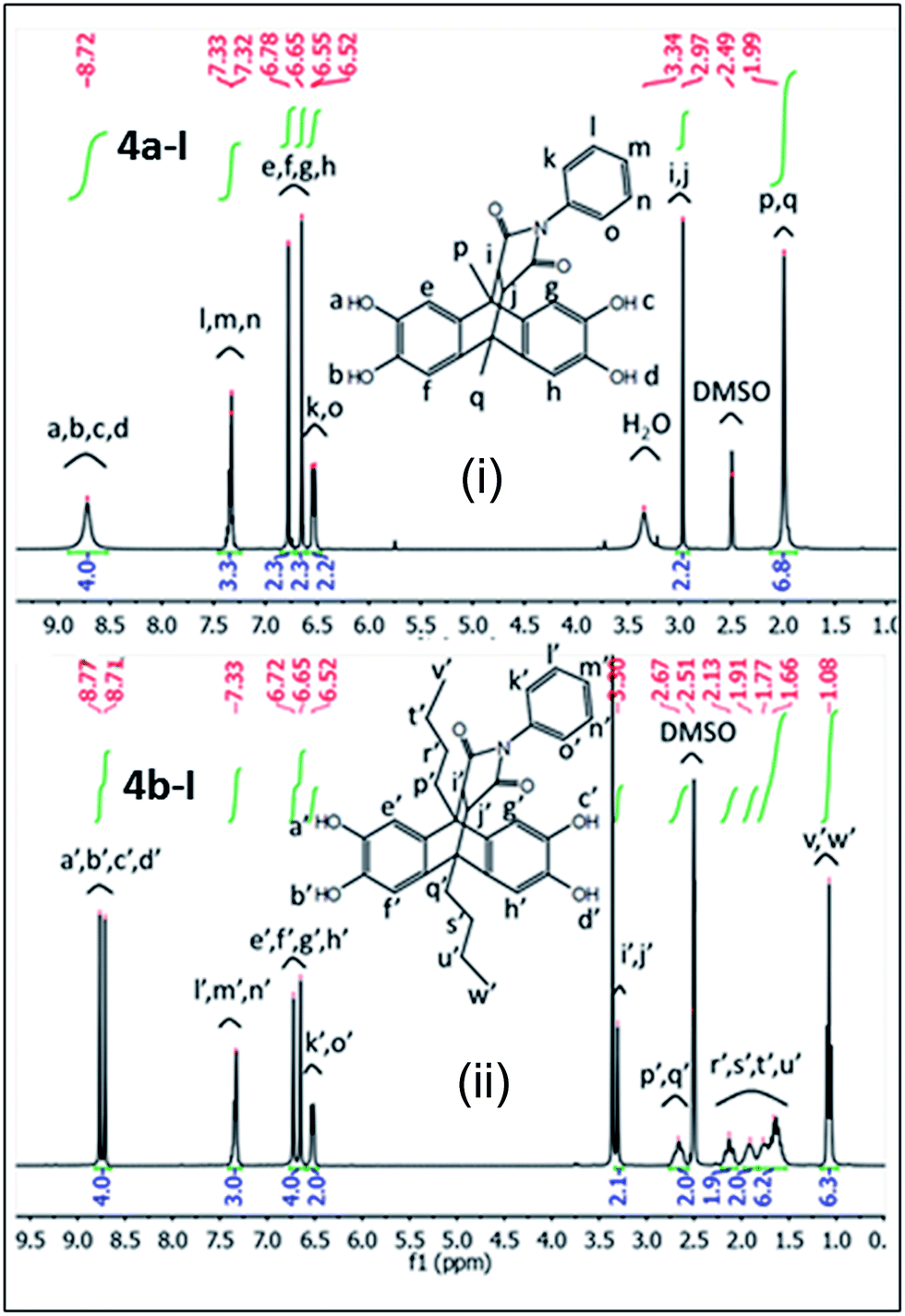 | ||
| Fig. 1 1H-NMR spectra of (i) 4a-1 and (ii) 4b-I. | ||
The other monomer pairs of 4a–b and the substituted anilines compare similarly in their analytical data. The monomer structures (4a-I and 4b-I) were also confirmed by FT-IR (ESI Fig. S1†). In general, the monomers on the basis of 4a are of low solubility, dissolving only in DMSO, DMF or DMAc, but not in low boiling solvents like chloroform, THF or acetone. The butyl groups, however, improve the solubility of monomers on 4b basis, allowing dissolution in chloroform or THF.
Generally, PIMs are obtained via polycondensation reaction at different temperatures; the original procedure of Budd/McKeown12 runs at 60 °C/3–4 days. Guiver et al.34 reduced the reaction time to few minutes by heating the reaction mixture to 155 °C and maintained stirrability by addition of toluene. We adopted the latter procedure to prepare PIM-1 and other PIMs but use diethyl benzene instead of toluene to keep the temperature inside the reaction mixture at 150 °C. The high temperature could cause the problem of a retro Diels–Alder reaction in the new series of monomers and therefore TGA measurements were conducted on 4a-I and 4b-I monomers to a thermal ramp up to 700 °C under argon atmosphere at a rate of 5 K min−1. By coupling with FT-IR the evolved gases were analysed. 4a-I proved to be stable at least till 280 °C and decomposition started at 295 °C. The 1st extract was measured at 334 °C, but a retro Diels–Alder with evaporation of phenylmaleimide was observed only at the 2nd extract at 358 °C. Fig. 2a shows the TGA mass loss analysis for 4a-I. Inset Fig. 2a shows the intensity of FTIR signals, which is connected to maximum gradient of weight loss in the TGA trace.
 | ||
| Fig. 2 TGA coupled with FTIR spectra of (a) 4a-1 and (b) 4b-I. | ||
Two extractions were carried out at two different temperatures (334 °C & 358 °C). Analysis of first extraction of gaseous decomposition product by FTIR spectra (Fig. 2 at 334 °C) shows a band at 2215 cm−1–2350 cm−1 and 3015 cm−1 (accompanied with low concentration of phenylmaleimide band also) which fitted to CO, CO2 and CH4 reference gas spectra from National Institute of Standard and Technology (NIST) Chemistry WebBook35 (see ESI for gas spectra of methane in Fig. S2†). The second extraction of gaseous decomposition at 358 °C shows a band at 575 cm−1, 1185 cm−1, 1360 cm−1 and 1785 cm−1, which fitted to reference gas spectra of phenylmaleimide35 (shaded region in inset Fig. 2a). Similarly, the TGA–FTIR measurement of 4b-I is depicted in Fig. 2b. The significant mass loss started at 250 °C and between 250–375 °C, approximately 60 wt% loss was observed in the compound, which corresponds to decomposition of 4b-I backbone. Analysis of first and second extraction of gaseous decomposition product by FTIR spectra at 334 °C and 358 °C shows a mixture of bands. The signals shows at 576 cm−1, 1184 cm−1, 1361 cm−1, 1786 cm−1 (for phenylmaleimide–shaded region inset Fig. 2(a and b)), 2215–2410 cm−1 (for CO2) and 3015 cm−1 (for C4H10), which fitted with the reference gas spectra of respective molecules (see ESI for gas spectra of butane Fig. S2†).35 In difference to 4a-I the retro Diels–Alder occurred already at lower temperature, in the 1st extract, however, stability till 250 °C was established. Therefore, the polymerisation at high temperature remains unaffected. The three dimensional (3D) view of TGA–FTIR of 4a-I and 4b-I are shown in ESI Fig. S3 and S4.†
3.2. Polymerization
The series of homopolymers and copolymers from the various monomers were synthesized via polycondensation reaction by reacting tetrahydroxy monomers with equimolar amounts of TFTPN, catalyzed by an excess of K2CO3, using a modified procedure similar to the synthesis of PIM-1 (Scheme 3).15,23–25,36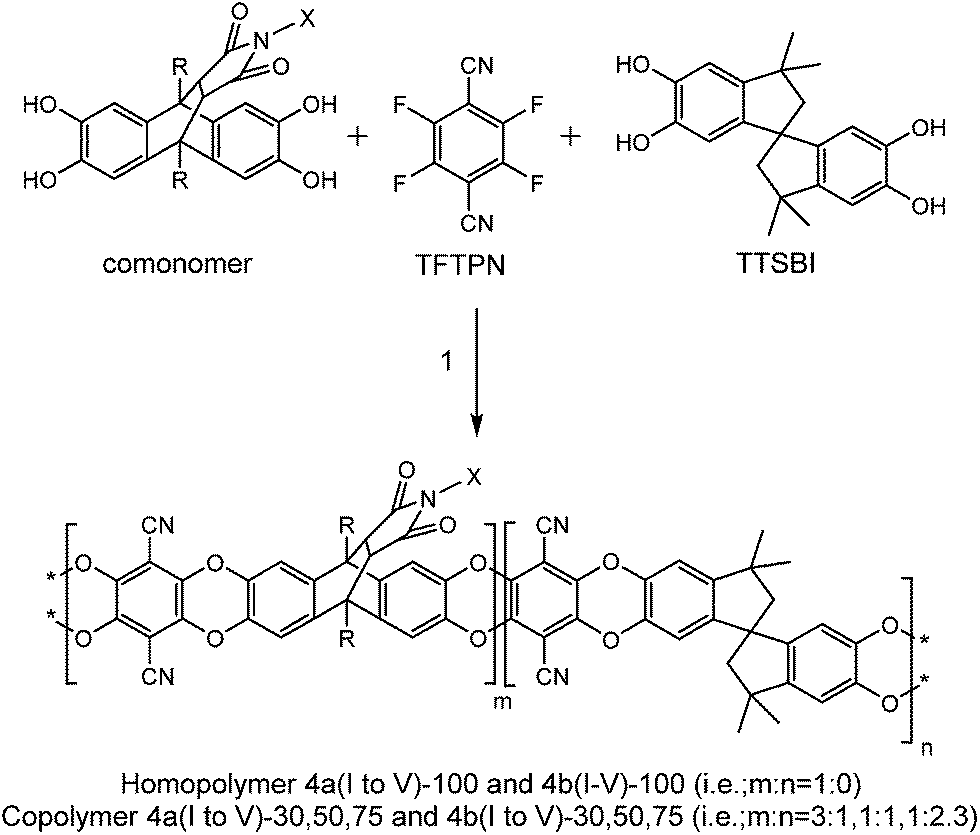 | ||
| Scheme 3 Synthesis of homopolymer and copolymers, 1–reagent and condition, K2CO3, DMAc (dimethylacetamide), DEB (diethylbenzene), 150 °C, 1/2 h. | ||
According to the mechanism of polycondensation reaction of poly(arylene ether)s, high temperature and high monomer concentration are favourable for increasing solubility of phenoxide salt and growing the polymer chain, hence, the occurrence of cyclic oligomers and crosslinked structure can be effectively reduced. In the present work, a slightly modified polymerization procedure15 was used compared to previously reported polymerization reaction.34 DEB was used in polymerization reaction instead of toluene (as described in procedure), which is advantageous not only to provide solubility enhancement of polymer but also provides a high boiling point solvent to the reaction.15,25 To keep the reaction mixture less viscous, additional amounts of DEB were added. The presence of DEB required precipitation of the polymers in methanol. The basic features of reaction and polymer characterization such as composition, molecular weight, thermal properties and copolymer composition estimated by 1H-NMR are listed in Table 1.
| Polymersa | Mwb (g mol−1) | Mnb (g mol−1) | PDI | TDc (°C) | Comonomer amountd (%) |
|---|---|---|---|---|---|
| a Each polymer is defined by the comonomer(s) from which they were made. For copolymers, the percentage of the contribution of comonomers to the TTSBI monomer (PIM-1 monomer) composition is given at the end of each polymer name.b From SEC analysis with calibrated polystyrene standards.c Actual onset temperature of decomposition.d Anthracene maleimide commoner-estimated by 1H-NMR.e n.s means that the polymer is insoluble in an appropriate solvent for SEC (i.e.CHCl3).f Soluble in CHCl3 and THF.g Primary onset temperature of decomposition.h Secondary onset temperature of decomposition. | |||||
| PIM-4aI-100 | n.se | n.s | — | 322 | — |
| PIM-4aI-50f | 25500 |
10200 |
2.5 | 311 | 42 |
| PIM-4aI-30f | 97700 |
39100 |
2.5 | 295 | 25 |
| PIM-4aII-100 | n.s | n.s | — | 321 | — |
| PIM-4aII-50f | 35900 |
13300 |
2.7 | 321 | 45 |
| PIM-4aII-30f | 110700 |
26400 |
4.2 | 327 | 30 |
| PIM-4aIII-100f | 19100 |
10600 |
1.8 | 337 | — |
| PIM-4aIII-50f | 35100 |
2500 | 13.8 | 347 | 43 |
| PIM-4aIII-30f | 76800 |
7500 | 10.2 | 368 | 26 |
| PIM-4aIV-100 | n.s | n.s | — | 326 | — |
| PIM-4aIV-50f | 63100 |
18000 |
3.5 | 347 | 48 |
| PIM-4aIV-30f | 67200 |
18700 |
3.6 | 317 | 30 |
| PIM-4aV-100 | n.s | n.s | — | 345 | — |
| PIM-4bI-100f | 111000 |
18500 |
6 | 266g, 440h | — |
| PIM-4bI-75f | 99900 |
32200 |
3.1 | 268g, 445h | 75 |
| PIM-4bI-50f | 198400 |
45100 |
4.4 | 273g, 450h | 48 |
| PIM-4bI-30f | 89000 |
21700 |
4.1 | 278g, 475h | 29 |
| PIM-4bII-100f | 95400 |
19900 |
4.8 | 279g, 445h | — |
| PIM-4bII-75f | 59500 |
22900 |
2.6 | 274g, 449h | 70 |
| PIM-4bII-50f | 31500 |
8500 | 3.7 | 283g, 459h | 51 |
| PIM-4bII-30f | 38450 |
12000 |
3.2 | 278g, 480h | 33 |
| PIM-4bIII-100f | 80200 |
19100 |
4.2 | 285g, 435h | — |
| PIM-4bIII-75f | 93300 |
24600 |
3.8 | 292g, 443h | 72 |
| PIM-4bIII-50f | 147300 |
23800 |
6.2 | 291g, 451h | 48 |
| PIM-4bIII-30f | 118700 |
22000 |
5.4 | 295g, 467h | 32 |
| PIM-4bIV-100f | 61800 |
28100 |
2.2 | 283g, 441h | — |
| PIM-4bIV-75f | 82100 |
19100 |
4.3 | 268g, 443h | 76 |
| PIM-4bIV-50f | 29300 |
5800 | 5 | 280g, 443h | 49 |
| PIM-4bIV-30f | 64000 |
10000 |
6.4 | 285g, 445h | 27 |
| PIM-4bV-100f | 153100 |
52800 |
2.9 | 278g, 436h | — |
| PIM-4bV-75f | 53400 |
11400 |
4.7 | 276g, 440h | 74 |
| PIM-4bV-50f | 59300 |
12600 |
4.7 | 289g, 443h | 48 |
| PIM-4bV-30f | 117000 |
16300 |
7.2 | 288g, 474h | 29 |
The average molar masses and polydispersities of homopolymers and copolymers were determined by SEC using polystyrene standards. For copolymers with methyl and butyl groups on the bridge head position [4a and 4b (I to V)] reasonable molecular weights with rather high polydispersity indices were obtained, which is consistent with the results of a typical polycondensation reaction. Table 1 also shows the copolymers composition, estimated from 1H-NMR spectra by integration of the peaks in the aromatic region (at ∼6.4 ppm) for TTSBI monomer (PIM-1 monomer) and aliphatic region for comonomer 4a and 4b (at ∼3.5 ppm). In each case the calculated composition reflected closely to the comonomer composition of the reaction (as an example shown in Fig. 3). Furthermore, the comparison of FT-IR spectra of copolymers shows a distinguishable increase of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration between 1660–1730 cm−1 with increasing content of comonomer 4a-I and 4b-I (ESI Fig. S5 and S6†). Differential scanning calorimetry (DSC) of all polymers showed no evidence of glass transition below the degradation temperature.
O stretching vibration between 1660–1730 cm−1 with increasing content of comonomer 4a-I and 4b-I (ESI Fig. S5 and S6†). Differential scanning calorimetry (DSC) of all polymers showed no evidence of glass transition below the degradation temperature.
 | ||
| Fig. 3 Comparative 1H-NMR spectra of copolymer with different composition derived from the comonomer (a) 4a-III and (b) 4b-III. | ||
Moreover, the homopolymers [PIM-CO4b(I–V)-100] derived from the new imido-monomers and TFTPN were synthesized under the same reaction conditions as PIM-1 (i.e. 150 °C and 30 min)34 and gave sufficiently high molecular weights. As the homopolymers obtained from monomers 4a (I–V) are insoluble in most organic solvents, the molecular weight determination by SEC was not possible. However, the homopolymers obtained from monomers 4b (I–V) exhibited good solubility in many organic solvents, e.g. chloroform and THF, due to the butyl groups on bridge head position, allowing both mechanically strong and flexible films to be cast. As an example, the images of homopolymer films derived from 4b (I–V) are shown in ESI Fig. S7.† Furthermore, thermo-gravimetric analysis (TGA) of homopolymers and copolymers derived from 4a (I–V) series showed that the onset of decomposition temperature is quite similar, while TGA of 4b (I–V) polymers and copolymers series showed two different onset decomposition temperatures (see ESI Fig. S8†). In case of 4b (I–V), the initial decomposition corresponds to decomposition of alkyl group along with maleimide derivatives and the second decomposition caused by breaking the backbone chain of the polymer.
We observed that the surface area of homopolymer (i.e. without any spiro unit) derived from the monomer [4a and 4b (I–V)] were in the range of 520–660 m2 g−1. The reason could be a greater cohesive interaction between the polymer chains due to the polar imido group, or occlusion of maleimide derivatives in the microporosity. The copolymers derived from the monomer (4b-III) were selected for further study. Table 2 gives BET surface area from nitrogen adsorption for copolymers with different ratio of monomer (4b-III) and also for the PIM-1 prepared under identical conditions in this study.
Fig. 4a compares the nitrogen adsorption/desorption isotherm at liquid nitrogen temperature (77 K) for PIM-4bIII-50 and PIM-1. Both polymers show high uptake at low relative pressure, a characteristic of microporous materials as defined by IUPAC (pore size < 2 nm).10 Both materials also demonstrated hysteresis, the desorption curve lying above the adsorption curve down to relative low pressures, a typical behaviour of microporous polymers that may be attributed to polymer swelling effects (p/p0 < 0.2).14 Analysis of the very low relative pressure regions of the adsorption isotherm by the Horvath–Kawazoe (HK) method gives the pore width distribution shown in Fig. 4b. Both polymers show a broad distribution of effective pore size in the micropore region (0.6–0.65 nm).
 | ||
| Fig. 4 (a) N2 adsorption (filled symbols) and desorption (open symbols) isotherm at 77 K for PIM-1 and PIM-4bIII-50 and (b) pore width distribution obtained by analysis of N2 adsorption at 77 K by the Horvath–Kawazoe (HK) method. | ||
From the literature,14 it was assumed that the roof-shaped structure once embedded within the polymer network possesses greater rigidity, which helps to enhance the microporosity. Comparing the triptycenecontaining A–B monomer precursor, it seems that the additional substituents on the maleimide unit make the polymer even more rigid but also fill some of the space created by the rigid polymer frameworks.
3.3. Gas separation performance
Free standing membranes of polymers for gas separation could be prepared by solution-casting and they were tough enough to allow measurement of gas permeability. Pure gas permeability coefficients (Pi) for H2, N2, O2, CO2, and CH4 were measured at 30 °C by time-lag instrument on dense polymer films of homopolymers [PIM-CO4b-100] and series of copolymers derived from monomers 4b(I–V). Gas permeation measurement was not possible for the homopolymer and copolymers obtained from the monomer 4a(I–V) because of their low molar masses leading to poor film forming properties. Permeabilities and ideal permselectivities (αij = Pi/Pj) are shown in ESI Tables S1 and S2,† respectively. All data were obtained from methanol treated membranes as mentioned in the experimental part. Immersing the membranes in methanol reverses the prior film formation history, in a manner similar to protocols previously developed for microporous polyacetylenes and PIM-1.38,39 For comparison, PIM-1 data of this present work and from literature are also included.40It is seen that the structure of anthracene maleimide derivatives exerts a strong effect on the observed permeability and permselectivity. Most homopolymers and the series of copolymers exhibit slightly higher selectivity, coupled with a reduction in gas permeability, compared to PIM-1. An exception is the series of copolymers of 4b-III, as they show comparable or slightly higher permeabilities than PIM-1 (shaded circle in Fig. 5).
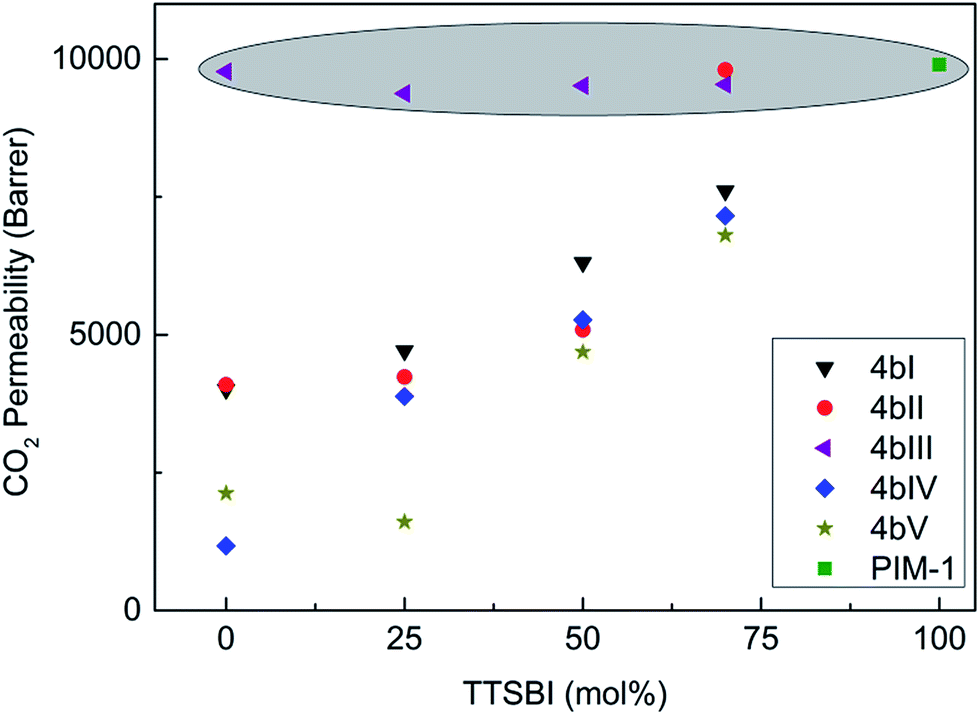 | ||
| Fig. 5 CO2 permeability of copolymers derived from monomer 4b (I to V) as a function of TTSBI content (mol%). | ||
For all other series of copolymer-PIMs (CO-PIMs), gas permeabilities were observed to increase with increasing ratios of TTSBI and therefore increasing similarity to PIM-1. PIM-CO4bIII-100 has higher CH4 and N2 permeability as PIM-1 and also the other copolymers with 4b-III behave similar to PIM-1. A reason for this should be the substitution of the aniline in o-position preventing rotation of the imido-group and consequently a close packing of the polymer chains. Also, the highly rigid three dimensional structures of anthracene maleimides 4a-III and 4b-III can be concluded on the 1H-NMR spectra showing three single peaks for the aromatic methyl groups at aniline. Another reason for comparable permeabilities could be the similar pore size distribution and higher surface area of copolymer 4b-III, shown in Table 2. For the other CO-PIMs, the p-substituted aniline is free in rotation and could be fitted in free volumes within the chain more easily leading to a reduction of permeability but increasing the selectivity.
The overall gas separation performances of newly synthesized polymers with comparison to Robeson's upper bound (1991 and 2008) for O2/N2, CO2/CH4 and CO2/N2 gas pairs are shown in Fig. 6.
 | ||
| Fig. 6 Robeson plot for (a) O2/N2 (b) CO2/CH4 and (c) CO2/N2 gas pairs showing data for methanol treated PIM-1 and CO-PIMs. The black and red lines represent the 1991 and 2008 upper bound, respectively. | ||
The performance of CO-PIMs for the gas pairs O2/N2 falls below the 2008 Robeson's upper bound. But for CO2/CH4 and CO2/N2 gas pairs, most of CO-PIMs show values near to upper bound 2008. However, for these gas pairs the selectivity is mainly due to the higher solubility dependent rather than diffusivity of CO2. Moreover, the polymers derived from newly synthesized monomers exhibit the competing behaviour to the promising spirobifluorene based PIM (SBF-PIM),41 thermally rearranged (TR) polymers42 polymers and tetrazole substituted PIMs (TZ-PIMs),43 that lie above the 2008 upper bound for CO2/CH4 and CO2/N2. Furthermore, the gas separation performance of CO-PIMs with comparison to upper bound (1991 and 2008) for other gas pairs such as H2/CH4, H2/CO2 and H2/N2 are shown in ESI Fig. S9.†
From an application point of view, temperature effects were studied on one of these copolymer membranes at higher temperature. Fig. 7 shows the permeability of N2, CH4 and CO2 for PIM-1 and PIM-CO4bIII copolymer membranes as a function of the inverse absolute temperature (temperature range 283–343 K, single gas at 1 bar feed pressure). Fig. 7 depicts that the permeability of the N2 and CH4 increased with increasing in temperature while for CO2 the permeability decreased with increasing temperature.
 | ||
| Fig. 7 Permeability of N2, CH4 and CO2 in PIM-1 and copolymers derived from the comonomer (4b-I) as a function of reciprocal temperature (●-PIM-4bIII-100, ▲-PIM-4bIII-75, ♦-PIM-4bIII-50, ▼-PIM-4bIII-30, ■-PIM-1). | ||
In order to understand the temperature dependence of N2, CH4, and CO2 permeabilities of these membranes, resulting data were correlated with the Arrhenius equation. Thus, the activation energy of permeation (Ep) was determined by the Arrhenius relationship:
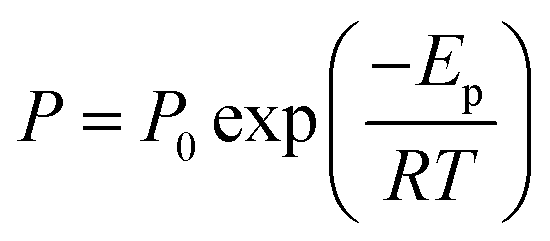 | (4) |
P is permeability, P0 is a pre-exponential factor, Ep is the activation energy of permeation (J mol−1), R is the gas constant (8.314 J (mol−1 K−1)) and T is the absolute temperature.
The activation energies of permeation (Ep) determined from the slope of the Arrhenius plot are shown in Table 3. It provides a qualitative measure of free volume and the size of molecular gaps between chain segments.44 In general, the activation energy of permeation is the sum of the activation energy of diffusion (Ed) and the enthalpy of sorption (ΔHs),
| Ep = Ed + ΔHs | (5) |
| Membranes | Ep (kJ mol−1) | ||
|---|---|---|---|
| N2 | CH4 | CO2 | |
| PIM-4bIII-100 | 3.5 | 3.4 | −9.8 |
| PIM-4bIII-75 | 4.1 | 3.8 | −6.4 |
| PIM-4bIII-50 | 4.7 | 5.2 | −8.2 |
| PIM-4bIII-30 | 3.5 | 5.0 | −8.0 |
| PIM-1 | 8.1 | 11.9 | −2.7 |
The activation energy of diffusion is a size-related parameter, its value depends on the size of the penetrant, the intra- and inter-molecular gaps between polymer chains, and the chain mobility. For activated diffusion processes, Ed is positive, meaning that diffusion coefficients increase with increasing temperature. The heat of sorption is related to the thermodynamic interaction of the penetrant with the polymer. Sorption of gases in polymers is typically an exothermic process. Thus, solubility of gases in polymers decreases with increasing temperature, and Hs is negative.
From Fig. 7, it was observed that gas permeability of N2 and CH4 for these membranes increases with increasing temperature, because Ed + Hs > 0 or |Ed|/|Hs| > 1, a typical behaviour of conventional glassy polymers. An exception to this rule is the temperature dependence of gas permeability in high-free-volume PIM-1 and its copolymer for condensable gases such as CO2, because gas permeabilities decrease with increased temperature (i.e. |Ed|/|Hs| < 1). This indicates that the effect of highly sorbed gases like CO2 does not affect the permeation rate of lighter gases in subsequent runs. The negative activation energies of permeation in high free volume glassy polymer result from very small activation energies of diffusion.45 Furthermore, negative activation energies of permeation are routinely observed for microporous solids in which the pore dimensions are relatively large in comparison with the diffusing gas molecules.44 On the other hand CH4 and N2 permeabilities of PIM-1 depend strongly on temperature and their Ep values are essentially of the same order of magnitude as those of conventional glassy polymers.
The long term stability tests for gases such as O2, N2 and CO2 were also carried out for two copolymers compositions derived from the monomer 4b-III (i.e. PIM-4b-III-50, PIM-4b-III-100). The copolymer was cast to thin film composite membranes on a PAN support and during the 800 h of the test these membranes were continuously flushed with N2 at 2 bar pressure. N2, O2 and CO2 permeances were measured at random over a period of 800 h. It was observed that the prepared copolymer composite membrane does not show any significant improvement in terms of permeance and selectivity O2/N2 and CO2/N2 gas pairs and behaves almost similar to PIM-1.
4. Conclusion
Ten new tetrahydroxy anthracene based monomers with imido-aniline substitution (4a,b I to V) were synthesized, characterized and employed in polycondensation reactions with TFTPN to form homopolymers and copolymers in combination with TTSBI. All synthetic procedures are easy, efficient and suitable for scale-up. The achieved PIMs were characterized by NMR, FT-IR, TGA, SEC and partly by BET surface area. The polymers obtained from monomers (4a) show low solubility in suited solvents such as CHCl3 and THF compared to polymers obtained from the monomer (4b). Changing the alkyl group (R) at the bridged anthracene from methyl to n-butyl group enhanced significantly the solubility. Therefore, polymers from monomer (4b) series could be cast into membranes and measured in single gas permeation of N2, O2, CO2 and CH4. Permeability of the new polymers is mostly lower or comparable to PIM-1, while selectivities are slightly higher or comparable to PIM-1. These results offer a variety of possible new monomers and polymer structures with potential in microporous materials for application in membrane technology and other areas where soft matters with large intrinsic free volume combined with chemical functionality are required.Acknowledgements
The authors would like to thank Silke Dargel and Petra Merten for technical support. This work was financially supported by Helmholtz Association of German Research Centres through the Helmholtz Portfolio MEM-BRAIN.Notes and references
- M. P. Tsyurupa and V. A. Davankov, React. Funct. Polym., 2002, 53, 193–203 CrossRef CAS.
- B. S. Ghanem, K. J. Msayib, N. B. McKeown, K. D. M. Harris, Z. Pan, P. M. Budd, A. Butler, J. Selbie, D. Book and A. Walton, Chem. Commun., 2007, 0, 67–69 RSC.
- N. B. McKeown, S. Hanif, K. Msayib, C. E. Tattershall and P. M. Budd, Chem. Commun., 2002, 0, 2782–2783 RSC.
- C. D. Wood, B. Tan, A. Trewin, H. Niu, D. Bradshaw, M. J. Rosseinsky, Y. Z. Khimyak, N. L. Campbell, R. Kirk, E. Stöckel and A. I. Cooper, Chem. Mater., 2007, 19, 2034–2048 CrossRef CAS.
- T. Masuda, E. Isobe, T. Higashimura and K. Takada, J. Am. Chem. Soc., 1983, 105, 7473–7474 CrossRef CAS.
- K. Nagai, T. Masuda, T. Nakagawa, B. D. Freeman and I. Pinnau, Prog. Polym. Sci., 2001, 26, 721–798 CrossRef CAS.
- K. Tanaka, M. Okano, H. Toshino, H. Kita and K.-I. Okamoto, J. Polym. Sci., Part B: Polym. Phys., 1992, 30, 907–914 CrossRef CAS.
- A. Y. Alentiev, V. P. Shantarovich, T. C. Merkel, V. I. Bondar, B. D. Freeman and Y. P. Yampolskii, Macromolecules, 2002, 35, 9513–9522 CrossRef CAS.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC.
- N. B. McKeown and P. M. Budd, Macromolecules, 2010, 43, 5163–5176 CrossRef CAS.
- M. Carta, K. J. Msayib, P. M. Budd and N. B. McKeown, Org. Lett., 2008, 10, 2641–2643 CrossRef CAS PubMed.
- P. M. Budd, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib and C. E. Tattershall, Chem. Commun., 2004, 230–231 RSC.
- D. Fritsch, P. Merten, K. Heinrich, M. Lazar and M. Priske, J. Membr. Sci., 2012, 401–402, 222–231 CrossRef CAS PubMed.
- T. Emmler, K. Heinrich, D. Fritsch, P. M. Budd, N. Chaukura, D. Ehlers, K. Rätzke and F. Faupel, Macromolecules, 2010, 43, 6075–6084 CrossRef CAS.
- D. Fritsch, G. Bengtson, M. Carta and N. B. McKeown, Macromol. Chem. Phys., 2011, 212, 1137–1146 CrossRef CAS.
- T. Defize, R. Riva, C. Jérôme and M. Alexandre, Macromol. Chem. Phys., 2012, 213, 187–197 CrossRef CAS.
- M. Helm, M. Petermeier, B. Ge, R. Fiammengo and A. Jäschke, J. Am. Chem. Soc., 2005, 127, 10492–10493 CrossRef CAS PubMed.
- M. Chandra and S. K. Silverman, J. Am. Chem. Soc., 2008, 130, 2936–2937 CrossRef CAS PubMed.
- K. T. Gagnon, S.-Y. Ju, M. B. Goshe, E. S. Maxwell and S. Franzen, Nucleic Acids Res., 2009, 37, 3074–3082 CrossRef CAS PubMed.
- H. Durmaz, A. Dag, A. Hizal, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7091–7100 CrossRef CAS.
- Z. Shi, J. Luo, S. Huang, Y.-J. Cheng, T.-D. Kim, B. M. Polishak, X.-H. Zhou, Y. Tian, S.-H. Jang, J. D. B. Knorr, R. M. Overney, T. R. Younkin and A. K. Y. Jen, Macromolecules, 2009, 42, 2438–2445 CrossRef CAS.
- X. Liu and J. K. Snyder, J. Org. Chem., 2008, 73, 2935–2938 CrossRef CAS PubMed.
- M. M. Khan, V. Filiz, G. Bengtson, M. M. Rahman, S. Shishatskiy and V. Abetz, Procedia Eng., 2012, 44, 1899–1901 CrossRef PubMed.
- M. Khan, V. Filiz, G. Bengtson, S. Shishatskiy, M. Rahman and V. Abetz, Nanoscale Res. Lett., 2012, 7, 504 CrossRef PubMed.
- M. M. Khan, V. Filiz, G. Bengtson, S. Shishatskiy, M. M. Rahman, J. Lillepaerg and V. Abetz, J. Membr. Sci., 2013, 436, 109–120 CrossRef CAS PubMed.
- J. G. Wijmans and R. W. Baker, J. Membr. Sci., 1995, 107, 1–21 CrossRef CAS.
- A. M. Shishatskii, Y. P. Yampol'skii and K. V. Peinemann, J. Membr. Sci., 1996, 112, 275–285 CrossRef CAS.
- M. M. Rahman, V. Filiz, S. Shishatskiy, S. Neumann, M. M. Khan and V. Abetz, Procedia Eng., 2012, 44, 1523–1526 CrossRef PubMed.
- M. M. Rahman, V. Filiz, S. Shishatskiy, C. Abetz, S. Neumann, S. Bolmer, M. M. Khan and V. Abetz, J. Membr. Sci., 2013, 437, 286–297 CrossRef CAS PubMed.
- A. Müller, M. Raltschewa and M. Papp, Ber. Dtsch. Chem. Ges. A, 1942, 75, 692–703 CrossRef.
- Y. V. Shklyaev and Y. V. Nifontov, Russ. Chem. Bull., 2002, 51, 844–849 CrossRef CAS.
- M. C. Kloetzel, in Organic Reactions, Wiley, 2011, pp. 1–59 Search PubMed.
- L. R. Dix, J. R. Ebdon, N. J. Flint, P. Hodge and R. O'Dell, Eur. Polym. J., 1995, 31, 647–652 CrossRef CAS.
- N. Du, J. Song, G. P. Robertson, I. Pinnau and M. D. Guiver, Macromol. Rapid Commun., 2008, 29, 783–788 CrossRef CAS.
- U.S. Department of Commerce.
- M. M. Khan, G. Bengtson, S. Shishatskiy, B. N. Gacal, M. Mushfequr Rahman, S. Neumann, V. Filiz and V. Abetz, Eur. Polym. J., 2013, 49, 4157–4166 CrossRef CAS PubMed.
- P. M. Budd, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib and C. E. Tattershall, Chem. Commun., 2004, 0, 230–231 RSC.
- A. J. Hill, S. J. Pas, T. J. Bastow, M. I. Burgar, K. Nagai, L. G. Toy and B. D. Freeman, J. Membr. Sci., 2004, 243, 37–44 CrossRef CAS PubMed.
- K. Nagai, L. G. Toy, B. D. Freeman, M. Teraguchi, T. Masuda and I. Pinnau, J. Polym. Sci., Part B: Polym. Phys., 2000, 38, 1474–1484 CrossRef CAS.
- P. M. Budd, N. B. McKeown, B. S. Ghanem, K. J. Msayib, D. Fritsch, L. Starannikova, N. Belov, O. Sanfirova, Y. Yampolskii and V. Shantarovich, J. Membr. Sci., 2008, 325, 851–860 CrossRef CAS PubMed.
- C. G. Bezzu, M. Carta, A. Tonkins, J. C. Jansen, P. Bernardo, F. Bazzarelli and N. B. McKeown, Adv. Mater., 2012, 24, 5930–5933 CrossRef CAS PubMed.
- H. B. Park, C. H. Jung, Y. M. Lee, A. J. Hill, S. J. Pas, S. T. Mudie, E. Van Wagner, B. D. Freeman and D. J. Cookson, Science, 2007, 318, 254–258 CrossRef CAS PubMed.
- N. Du, H. B. Park, G. P. Robertson, M. M. Dal-Cin, T. Visser, L. Scoles and M. D. Guiver, Nat. Mater., 2011, 10, 372–375 CrossRef CAS PubMed.
- I. Pinnau and L. G. Toy, J. Membr. Sci., 1996, 109, 125–133 CrossRef CAS.
- T. Masuda, Y. Iguchi, B.-Z. Tang and T. Higashimura, Polymer, 1988, 29, 2041–2049 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra03663h |
| This journal is © The Royal Society of Chemistry 2014 |