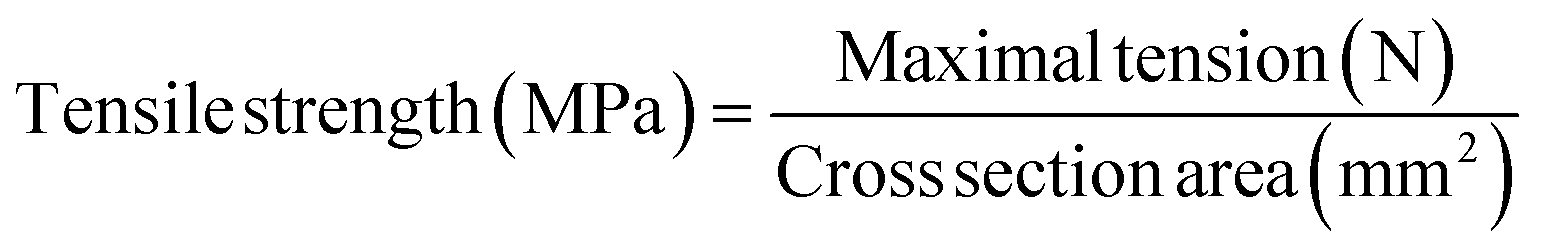DOI:
10.1039/C4RA03633F
(Paper)
RSC Adv., 2014,
4, 31400-31408
Cooperative fabrication of ternary nanofibers with remarkable solvent and temperature resistance by electrospinning†
Received
21st April 2014
, Accepted 2nd July 2014
First published on 2nd July 2014
Abstract
Novel ternary organic–inorganic composite nanofibrous mats are successfully prepared by cooperative fabrication via electrospinning. Poly(vinyl alcohol) (PVA) and poly(acrylic acid) (PAA) were chosen as the polymer components, while tetraethyl orthosilicate (TEOS) was used as the silicon source. By adjusting the mass ratio of polymer and silica gel, the obtained nanofibers with a mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.75 exhibit very uniform morphology with an average diameter of 845 nm. Systematic investigations were performed to illustrate the reaction process and internal structure of the nanofibers. The esterification and dehydration between PVA, PAA and hydrolytic TEOS are proposed to form an integrated network structure which makes the ternary electrospun nanofibrous mats more stable compared to pure single polymer and the binary composites. Eight typical solvent (H2O, DMF, EtOH, THF, MeOH, CHCl3, n-hexane and acetone) immersion experiments of the nanofibrous mats at varied temperatures (60, 80, 100, 120, 150 and 200 °C) were performed for the study of its solvent and thermal stability. The results show that the prepared ternary organic–inorganic nanofibrous mats exhibit remarkable solvent and temperature resistance (e.g. in DMF at 200 °C). Moreover, the obtained PVA/PAA/SiO2 nanofibrous mats also present desirable mechanical properties such as malleability with a high breaking elongation. Taking account of the availability of raw materials, easy maneuverability, and the outstanding chemical stability and mechanical properties, such a cooperative fabrication strategy could find great extensibility for the synthesis and application of multiple organic–inorganic composite materials.
:0.75 exhibit very uniform morphology with an average diameter of 845 nm. Systematic investigations were performed to illustrate the reaction process and internal structure of the nanofibers. The esterification and dehydration between PVA, PAA and hydrolytic TEOS are proposed to form an integrated network structure which makes the ternary electrospun nanofibrous mats more stable compared to pure single polymer and the binary composites. Eight typical solvent (H2O, DMF, EtOH, THF, MeOH, CHCl3, n-hexane and acetone) immersion experiments of the nanofibrous mats at varied temperatures (60, 80, 100, 120, 150 and 200 °C) were performed for the study of its solvent and thermal stability. The results show that the prepared ternary organic–inorganic nanofibrous mats exhibit remarkable solvent and temperature resistance (e.g. in DMF at 200 °C). Moreover, the obtained PVA/PAA/SiO2 nanofibrous mats also present desirable mechanical properties such as malleability with a high breaking elongation. Taking account of the availability of raw materials, easy maneuverability, and the outstanding chemical stability and mechanical properties, such a cooperative fabrication strategy could find great extensibility for the synthesis and application of multiple organic–inorganic composite materials.
Introduction
The electrospinning technique is a simple, effective and versatile method to produce a wide variety of fibrous materials such as polymers, ceramics and composites.1–5 The properties of the materials could be easily tuned either by varying the system parameters (e.g., the viscosity and dielectric properties of the spinning solution) or by the process parameters (e.g., electric potential, flow rate and concentration, distance between the capillary and collection screen).6,7 Electrospun nanofibrous materials have attracted great attention due to their relatively large specific surface area to volume ratio, flexibility in surface functionalities and high production rates. These materials have found a wide range of applications including tissue engineering, filtration, sensing, catalysis, drug delivery and adsorption.8–12
For the environmental concerns and industrial production demands, electrospinning from the aqueous or alcoholic solutions is highly recommended. So far, fibrous structures of polymers, especially those biocompatible, cost-effective polymers, such as poly(vinyl alcohol) (PVA), poly(acrylic acid) (PAA), poly(methyl methacrylate) (PMMA) and poly(vinylpyrrolidone) (PVP) have been produced under the aqueous or alcoholic conditions using the electrospinning technique. These polymers possess hydrophilic functional groups which contribute to their versatile physical and chemical properties, and provide great interfaces for further modification. However, the high hydrophilicity could lead to the disintegration of the fiber once it contacts to water or other polar solvents upon certain conditions, which greatly limits the application of these polymer fibers. In order to reduce the undesired hydrophilicity and enhance the stability against water, self-crosslinking or crosslinking of multicomponent polymers through chemical or physical approaches has been widely used. Baştürk et al.13 prepared water insoluble PVA/PAA nanofiber mats by cross-linking the polymers via heat-induced esterification. Li et al.14 prepared ultra-fine fibrous membranes with different ratio of PVA/PAA and studied the swelling ratio. There is no doubt that the water-stability of electrospun polymer nanofibers can be enhanced by the inter-polymer crosslinking. However, most of these products are thermally unstable and poor-resistant to organic solvents.15–17 Organic–inorganic nanocomposites materials possess both the merits of organic polymers and inorganic materials,18 combining the compatibility, paintability and flexibility with high strength, thermal stability and solvent resistance. Hence, organic–inorganic composite materials are more promising for utilization in harsh conditions. There have been some reports on preparing organic–inorganic materials by electrospinning technique combined with the sol–gel methodologies.19,20 Among them, hydrophilic polymer–silica hybrid nanofiber materials are the most studied composites. These materials are based on the dehydration between the hydrolyzed silane and the carboxylic acid or hydroxyl groups of the polymer to form three-dimension network which exhibit great thermal and chemical stability. Shao et al.16 produced poly(vinyl alcohol)–silica composite fiber mats and studied the swelling property; Pirzada et al.21 prepared hybrid silica–PVA nanofibers with different silica–PVA ratio and aging time of the silica precursor mixture; Xu et al.22 prepared mesoporous poly(acrylic acid)/SiO2 composite and studied the sorption behaviour of malachite green. Although there are some works reported on the synthesis of polymer–silica hybrid composites, only single polymer is selected as the organic template and few systematic studies are conducted on the stability in organic solvents at high temperatures and their mechanical properties, which is fundamentally crucial for the development of functional materials with great chemical stability. We have reported the preparation of hybrid PVA–silica fibrous membrane via the crosslinking between PVA and inorganic silane using the electrospinning technique and was successfully used as the sorbents for the removal of aqueous contaminants.12 Although the product was stable in water, the membrane still showed quite poor stability at high temperature and in organic solvents. Recently, Xu et al.23 reported that using a copolymer of poly[styrene-co-3-(trimethoxysilyl)propyl-methacrylate] in the electrospun nanofibrous mats could improve the solvent and temperature resistance. However, the costly silane raw materials and the complicated synthetic procedure hinder its large-scale application.
In order to further improve the stability of nanofibers in organic solvents and at high temperatures, here we report the preparation of a ternary composite PVA/PAA/SiO2 nanofibrous mats via the electrospinning technology. The two polymers are both cross-linked to the silica content to form an integrated network structure. Systematic investigations are then performed to understand the internal structure of the nanofibers and how the network structure improves the solvent and temperature resistance, as well as the mechanical properties. Since the net structure was formed between the polymer and silica content, the PVA/PAA/SiO2 nanofibrous mats showed remarkable resistance to typical solvents and high temperatures. At the same time, their mechanical properties turned out to be great. Results from this study suggest that cost-effective raw materials availability, easy maneuverability and outstanding solvent and temperature resistance performance render this approach much expectable in the preparation of electrospun multi-functional organic–inorganic composite materials.
Experimental section
Materials
Poly acrylic acid (PAA, Mw = 450000) was purchased from Sigma-Aldrich. Poly vinyl alcohol (PVA, Mw = 77000), tetraethyl orthosilicate (TEOS), hydrochloric acid (HCl), N,N-dimethylformamide (DMF), ethanol (EtOH), tetrahydrofuran (THF), methanol (MeOH), trichloromethane (CHCl3), n-hexane and acetone were provided by Sinopharm Chemical Reagent Company, China. All reagents were of analytical grade and used without further purification. Deionized (DI) water was used throughout this work.
Preparation of PVA/PAA/SiO2 gel
Firstly, PVA powder and PAA particles with molar composition 1:1 were mixed and dissolved in DI water to obtain a 5 wt% polymer gel (specifically, 0.26 g PVA and 1.5 g PAA were dissolved in 33.38 g DI water). The mixture was kept stirring for 6 h in a water bath at 80 °C to form a polymer gel. Silica gel was prepared by mixing 2 g EtOH and 2.5 g TEOS followed by adding 0.1 mL of 1 mol L−1 HCl as a catalyst. The silica gel was stirred for 15 min at room temperature. Finally, 0.5 g silica gel and 2 g polymer gel were mixed and kept in a water bath at 70 °C for 15 min. The mixture was kept stirring until the gel cooled to room temperature, and thus a viscous gel of PAA/PVA/SiO2 composite was obtained and recorded as G-1. Three more composites G-2, G-3 and G-4 with the mass ratio (silica gel:polymer gel) of 0.5, 0.75, 1 respectively were prepared in the same way, and G-0 was the pure polymer gel (Table 1).
Table 1 The components of raw materials used in the synthesis of PVA/PAA/SiO2 composites
| |
Polymer gel |
Silica gel |
| PVA (g) |
PAA (g) |
Water (g) |
TEOS (g) |
EtOH (g) |
HCl (mL) |
| G-0 |
0.086 |
0.5 |
11.13 |
0 |
0 |
0 |
| G-1 |
0.086 |
0.5 |
11.13 |
1.63 |
1.30 |
0.065 |
| G-2 |
0.086 |
0.5 |
11.13 |
3.26 |
2.60 |
0.13 |
| G-3 |
0.086 |
0.5 |
11.13 |
4.89 |
3.90 |
0.195 |
| G-4 |
0.086 |
0.5 |
11.13 |
6.52 |
5.20 |
0.26 |
Preparation of ternary electrospun nanofibers
The electrospinning gel that consists of polymer gel with or without silica gel was transferred to a plastic syringe and ejected from a stainless steel needle at a rate of 1.0 mL h−1. The stainless steel needle served as an electrode, while a glass plate covered with aluminium foil was used as the counter electrode. A DC voltage of 20 kV was applied to the needle and the distance from the needle to the collector was 14 cm. A dense web of fibers was collected on the aluminium foil and then removed from the foil and dried at 140 °C for 15 min to get the ternary electrospun nanofibers. The samples were recorded as N-0, N-1, N-2, N-3 and N-4 for films prepared from G-0, G-1, G-2, G-3 and G-4 gels. The obtained composite nanofibers were kept in a desiccator for further analysis.
Characterization
The molecular structure of the nanofibers was characterized by Fourier transform infrared spectroscopy (FTIR) of KBr powder pressed pellets and was recorded on a Bruker Vector 22 spectrometer. X-ray diffraction (XRD) patterns of the samples were recorded using Bruker D8 Advance X-ray powder diffractometer at 40 kV and a current of 40 mA. Thermal Gravimetric Analysis (TGA) was performed on TA Instrument TGA-7. Typically, 5–10 mg of sample was placed in an alumina pan under air flow (100 mL min−1) and heated at a rate of 10 °C min−1 from room temperature to 900 °C. Scanning electron microscopy (SEM) measurements were taken on a JEOL-2100F field emission scanning electron microscopy. Nitrogen adsorption–desorption measurements were conducted with Micro-meritics ASAP2020M analyzer at 77 K. The sample was degassed at 150 °C for 6 h in the vacuum line before the tests. The surface topographies of the nanofibers were examined using a Dimension 3100 atomic force microscope (AFM) in the tapping mode and using a silicon tip.
Stability studies
The electrospun nanofibrous mats were immersed into different solvents to analyse the solvent resistance. In this study, 8 different solvents were chosen including H2O, DMF, EtOH, THF, MeOH, CHCl3, n-hexane and acetone. These solvents are typical polar solvents except n-hexane. As far as temperature was concerned, the experiments were conducted at 60, 80, 100, 120, 150 and 200 °C depending on the boiling point of the solvents (Table S1†). The nanofibrous mats were put into airtight containers and all the experiments were carried out in oven.
Mechanical property tests
Mechanical property tests of the electrospun nanofibrous mats were carried out using a universal testing machine (FR-103C) equipped with 5 kN load cell and mechanical grips. The samples were cut into dumbbell shape (Fig. S1†) by cut-off knife. The specific size was measured by vernier caliper while the thickness was measured by thickness gauge. Tensile tests were performed in displacement controlled mode with loading rate of 10.0 mm min−1. Tensile strength was calculated by maximal tension and cross-section area of the films (eqn (1)), while yield strength was calculated by the tension at yield (eqn (2)). Breaking elongation was counted by the displacement at the breaking and the scale distance (eqn (3)).| |
 | (1) |
| |
 | (2) |
| |
 | (3) |
Results and discussions
Characterization of composite nanofibers
The FTIR spectra of the as prepared ternary nanofibers are shown in Fig. 1 which provide information on the types of chemical bonding.21 The broad feature observed at around 3400 cm−1 could be attributed to the O–H stretching vibration of the hydroxyl group from PVA or the hydrolyzed TEOS.24 With the increase of silica content from N-1 to N-4, this peak becomes more and more obvious. The vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond from PAA polymer appears at 1717 cm−1 for sample N-0.25 The same peak is also observed at the same wavenumber for N-1, N-2, N-3 and N-4 but with much less intensity. The peak at 943 cm−1 appears for samples N-1, N-2, N-3 and N-4, which is assigned to Si–OH stretching vibration. The peak at 1654 cm−1 is also assigned to Si–OH vibration, which is only observed for samples N-3 and N-4 with high SiO2 content.21 The characteristic vibration peaks of Si–O–Si bond are clearly seen around 1083 cm−1, 800 cm−1 and 461 cm−1 which give the evidence of the silica network formation.26,27 In addition, the intensities of Si–OH and Si–O–Si peaks increase from N-1 to N-4 indicating the increase of SiO2 content in the composite nanofibers. It is worth noting that there is a shoulder on the Si–O–Si peak around 1110 cm−1 which is attributed to the Si–O–C bonds.21 The presence of this bond in the nanofiber is important as it gives evidence that there are new chemical bonding formed between the polymer and silica backbone. The results indicate that the three components are not just physically mixed but are more possibly integrated into a network via chemical bonding.
O bond from PAA polymer appears at 1717 cm−1 for sample N-0.25 The same peak is also observed at the same wavenumber for N-1, N-2, N-3 and N-4 but with much less intensity. The peak at 943 cm−1 appears for samples N-1, N-2, N-3 and N-4, which is assigned to Si–OH stretching vibration. The peak at 1654 cm−1 is also assigned to Si–OH vibration, which is only observed for samples N-3 and N-4 with high SiO2 content.21 The characteristic vibration peaks of Si–O–Si bond are clearly seen around 1083 cm−1, 800 cm−1 and 461 cm−1 which give the evidence of the silica network formation.26,27 In addition, the intensities of Si–OH and Si–O–Si peaks increase from N-1 to N-4 indicating the increase of SiO2 content in the composite nanofibers. It is worth noting that there is a shoulder on the Si–O–Si peak around 1110 cm−1 which is attributed to the Si–O–C bonds.21 The presence of this bond in the nanofiber is important as it gives evidence that there are new chemical bonding formed between the polymer and silica backbone. The results indicate that the three components are not just physically mixed but are more possibly integrated into a network via chemical bonding.
 |
| | Fig. 1 FTIR spectra of the PVA/PAA/SiO2 ternary nanofibers with different mass ratio of polymer and silica gel. | |
The reasons for choosing PVA and PPA as polymers are as followed. PVA and PAA are hydrosoluble polymers which are widely used in the electrospinning process with great performance in the formation of nanofibers.3,28,29 Both of them can be dissolved in water to form gel with different viscosity, meanwhile the hydroxyl group of PVA and the carboxyl of PAA could react with other chemicals.
Based on the FTIR results, a possible reaction process and structural model of the PVA/PAA/SiO2 composite nanofibers is proposed and illustrated in Fig. 2. The reactions might occur in three main aspects: (1) the self-condensation reaction of the silanol groups;30,31 (2) dehydration reaction between the hydroxyl group of PVA and TEOS hydrolysate; (3) esterification reaction between the hydroxyl of TEOS hydrolysate and the carboxyl of PAA.24,26 There are a large amount of hydroxyl groups in the hydrolysed TEOS and they can undergo self-condensation to form silica. The surplus hydroxyl react with PVA and PAA to form a network structure consisting of carbon chain and silica skeleton. As to previous report, this network formation could greatly improve the solvent and temperature resistance of the composite nanofibers.
 |
| | Fig. 2 The proposed structural model of the PVA/PAA/SiO2 ternary nanofibers. | |
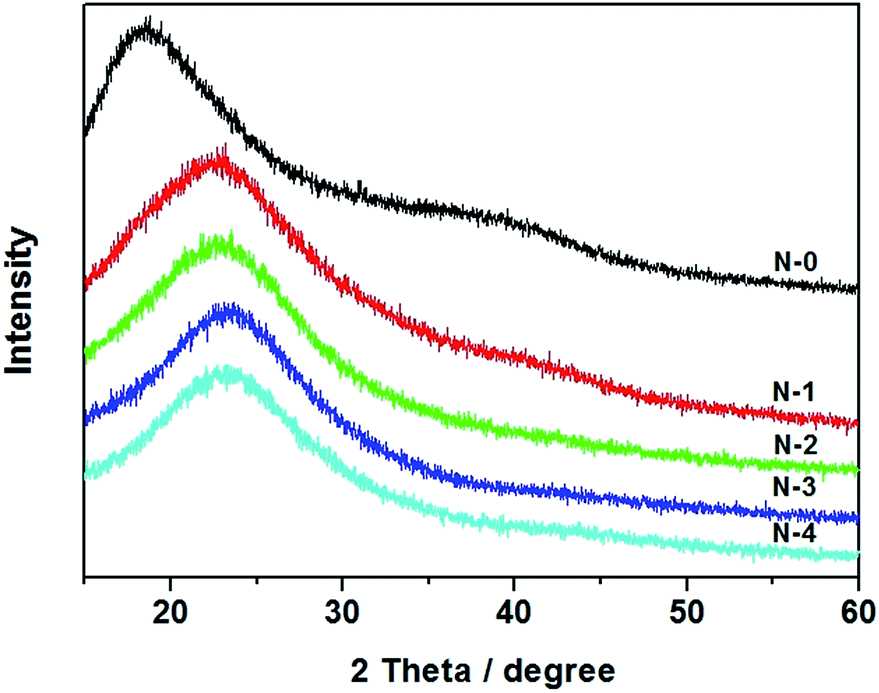
Wide-angle XRD was performed to examine the crystalline nature of the PVA/PAA/SiO2 composite nanofibers (Fig. 3). For PAA/PVA nanofibers without silica (N-0), only a wide peak is observed at around 2θ = 19° which is in agreement with the XRD pattern of the crystalline pure PVA nanofibers.32 The result indicates that PAA/PVA nanofibers possess a semi-crystalline structure.33,34 When TEOS was added to the electrospinning gel, the position and intensity of the peak changed. Specifically, the XRD peak moved to around 2θ = 24° which gives evidence that the added SiO2 might interfere with PAA/PVA resulting in the structure change.35 It is also noticed that with increasing content of TEOS from N-1 to N-4, there is not much change to the XRD peak, suggesting the crystalline structure of the composites remain the same and is not affected by the silica ratio.
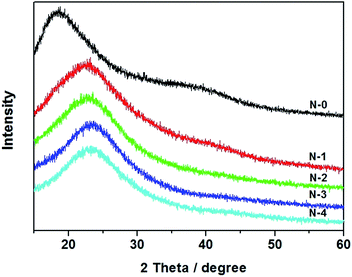 |
| | Fig. 3 XRD patterns of the PVA/PAA/SiO2 ternary nanofibers with different mass ratio of polymer and silica gel. | |
The degradation behaviour of PVA/PAA/SiO2 ternary nanofibers was measured by the thermogravimetric analysis (TGA) in air flow (Fig. 4). For sample N-0, the major weight loss starts at 200 °C due to the cleavage of C–C bonds of the polymer and the components turn into CO2 and water.21 Nearly complete degradation of the polymer is observed at approximately 450 °C and the residues was only account for 3.12% by weight.36 The major weight loss of PVA/PAA/SiO2 composite nanofibers occurs in the temperature range from 200 °C to 520 °C and is mainly attributed to the degradation of polymer content. The TGA curves show that with the increasing TEOS content in the electrospun gel (from N-1 to N-4), the weight decrease was slower, with total weight losses of 60.36%, 42.44%, 31.9% and 27.29% respectively. These values correspond to the silica gel ratios in the composites were 0.25, 0.5, 0.75 and 1 respectively. It is also noticed in Fig. 4 that compared with the composite nanofibers, the pure polymer fibers (N-0) had less weight loss at the initial stage of the heat program (100–220 °C). This might be due to that the hydrogen bonding within PVA/PAA molecules is stronger than that in the PVA/PAA/SiO2 composite nanofibers.21 According to the above results, the introduction of TEOS clearly enhances the thermal stability of the composite nanofiber membranes.
 |
| | Fig. 4 TGA curves of the PVA/PAA/SiO2 ternary nanofibers with different mass ratio of polymer and silica gel. | |
Nanofibers of PVA/PAA are water soluble due to the polar functional groups of carboxyl and hydroxyl groups.37,38 Previous studies have found that PVA/PAA nanofibers become insoluble in water after thermal treatment for the reason that cross-linking reaction (esterification) was induced by heat between the carboxyl group in PAA and the hydroxyl group in PVA.13,14,37 In this study, the esterification was achieved within the nanofibers containing equal molar ratio of PVA and PAA by conducting the thermal treatment at 140 °C for 15 min.13,14,39 As to SEM measurement, the cross-linked PVA/PAA nanofibers remain the nanofibrous structure (Fig. S2a†) with a mean diameter of 329 nm (Fig. S2d†). It is also notable that the nanofibrous mats become rigid after the thermal treatment. After immersing the nanofibers in H2O, DMF, EtOH and MeOH for 24 h (Fig. S2b, c, e and f†), though the nanofibrous structure was basically retained, the nanofibers were metamorphotic and swollen to some extent. The degree of swelling was more obvious in water than in other three solvents. The possible explanation is that some carboxyl and hydroxyl groups were unreacted in the cross-linking procedure, which could interact with the solvents resulting in the swelling.15,40
In this work, the main electrospinning parameters considered were voltage and distance between needle and target. Both factors influenced the electrospinning process significantly, especially on the morphology and diameter of the fibers. Through a series of studies, the optimal electrospinning parameters turned out to be 20 kV and 14 cm. The details were listed in the ESI (Fig. S3†). On the other hand, the selection of solvent in the preparation of the electrospinning gel was also important. PVA and PAA are hydrosoluble polymers and the viscosity of the solution increased with the increasing concentration. The electrospinning solution should possess certain viscosity in order to form uniform fibers. TEOS hydrolyzed very quickly in water. Adding EtOH into the solution could make the process more controllable.
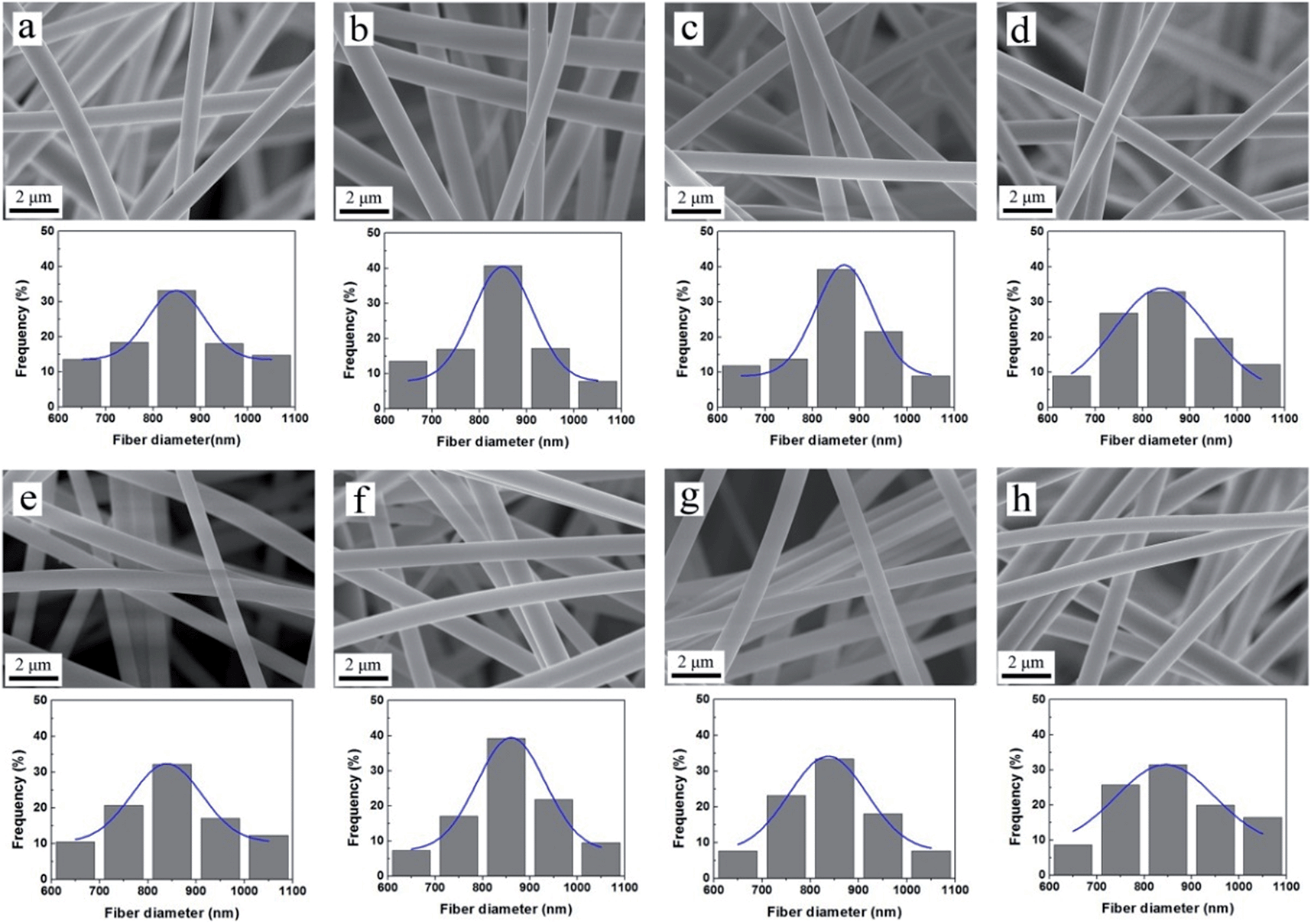
Fig. 5 shows SEM images of the nanofibers prepared by different mass ratios of silica gel and polymer gel. It is notable that electrospinning parameters and particular solvent have influences in the experiments. With increasing amount of silica gel from G-1 to G-4, the diameters of composite nanofibers become larger (Fig. 5a–d)21,26 since silica gel generates viscous silica network.35,41 We notice that fibers of sample N-1 and N-2 show some cross sections and are not well-distributed; N-4 sample shows obvious beads and the diameter of one single fiber fluctuates. Among the four samples, nanofibers of sample N-3 exhibit outstanding uniform morphology (Fig. 5c, e and f) and smooth surface (Fig. 5h). The diameters of the fibers ranged from 619 to 1087 nm, and the mean diameter is 845 nm (Fig. 5g).
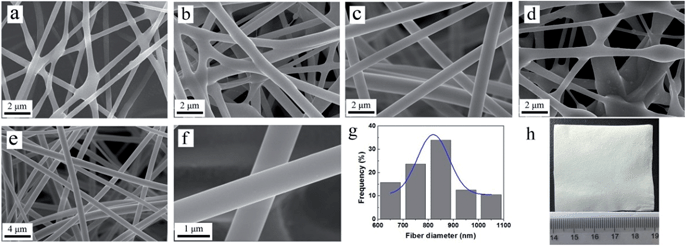 |
| | Fig. 5 SEM images of the as prepared (a) N-1 (b) N-2 (c) N-3, (d) N-4 composite nanofibers; (e and f) sample N-3 at different magnifications; (g) sample N-3: the statistical distribution of fiber diameters and its Gaussian fit. (h) Optical image of sample N-3. | |
Nitrogen adsorption–desorption measurement was conducted to study the porosity of the prepared composite nanofibers. Sample N-3 has a specific surface area and pore volume of 1.72 m2 g−1 and 0.003 cm3 g−1, respectively (Fig. S4†) which show that the nanofibers were non-porous and the surface area could be attribute to the external surface of the fibers.
Study on the solvent resistance
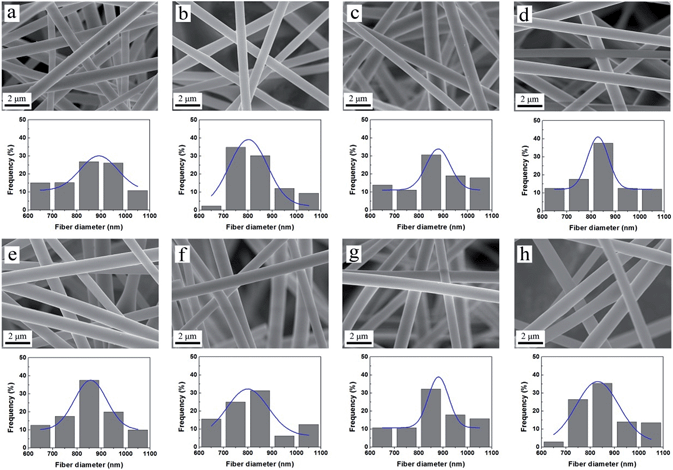
To study the solvent resistance of the composite nanofibers, sample N-3 was immersed in various solvents including H2O, DMF, EtOH, THF, MeOH, CHCl3, n-hexane and acetone at room temperature for 24 h (Fig. 6a–h).23 All the solvents are polar except for n-hexane. Neither deformation nor fracture is observed in the SEM images and the surfaces of the nanofibers remain smooth. After another 24 h immersion, the morphology and structure were still kept intact (Fig. S5†). Most nanofibers are in the range between 800 and 900 nm and the Gauss fitting curve shows a normal distribution for the two groups. The mean diameters of the fibers after 24 h immersion vary between 833 and 879 nm (Fig. S6a†); while the figures for 48 h group are 843 to 876 nm (Fig. S6b†). These indicated that there was very little swelling occurred for nanofibers in all solvents studied, either polar or non-polar solvents. This remarkable resistance to the most common solvents should be attributed to the network structure formed between the polymer and silica which greatly enhances the stability of the composite nanofibers.
 |
| | Fig. 6 SEM images of sample N-3 immersed in (a) H2O, (b) DMF, (c) EtOH, (d) THF, (e) MeOH, (f) CHCl3, (g) n-hexane, (h) acetone for 24 h at room temperature. The figure under the SEM image shows the corresponding statistical distribution of fiber diameters and its Gaussian fit. | |
Temperature resistance of composite nanofibers
In order to study the effect of temperature, the experiments were carried out at 60 and 80 °C, in which H2O, DMF, EtOH and n-hexane and were used as the representative solvents.23 SEM images illustrated that compared with the room temperature control group (Fig. 6a–c and g), the morphology of the fibers keeps unchanged after 24 h immersion (Fig. 7). Both the average diameter and diameter distribution of the 60 and 80 °C group show little difference. Thus the temperature at 60 and 80 °C hardly affect the stability and structure of the composite nanofibers. Then the nanofibers were immersed in H2O, DMF and EtOH at 100 °C for 24 h. The obtained images (Fig. 8a–c) reveal intact structures of the nanofibers. When the test temperature increased to 120 °C, some of the fibers fractured and the structure were damaged to a little extent (Fig. 8d and e). The results could be attributed to the fact that the test temperature was higher than the boiling point of H2O (100 °C) and EtOH (78 °C). Once the solvents reach boiling, the pressures in the airtight container are too high to destroy the nanofibers. For DMF with a boiling point of 153 °C, the measurements were conducted at 150 °C and 200 °C to study the resistance of high temperature (Fig. 8f and g). Even the temperature of 200 °C exceeded the boiling point of DMF, the nanofibrous mat remain undamaged after immersion for 24 h (Fig. 8h). The composite nanofibers thus have excellent stability in DMF solvent at a temperature as high as 200 °C.
 |
| | Fig. 7 SEM images of sample N-3 immersed in (a) H2O 60 °C, (b) DMF 60 °C, (c) EtOH 60 °C, (d) n-hexane 60 °C, (e) H2O 80 °C, (f) DMF 80 °C, (g) EtOH 80 °C, (h) n-hexane 80 °C for 24 h. The figure under the SEM image shows the corresponding statistical distribution of fiber diameters and its Gaussian fit. | |
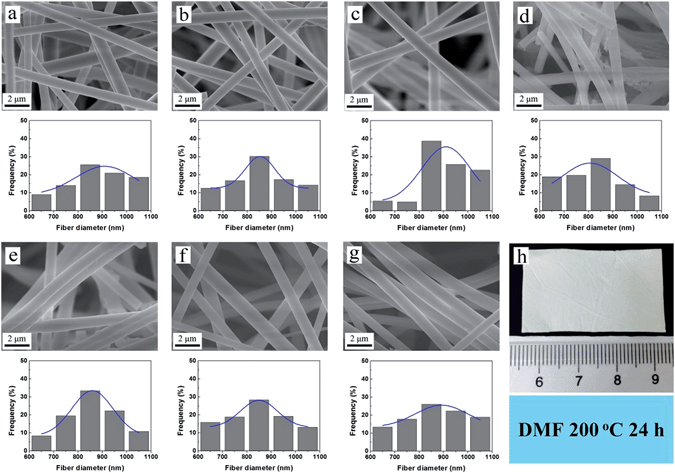 |
| | Fig. 8 SEM images of sample N-3 immersed in (a) H2O 100 °C, (b) DMF 100 °C, (c) EtOH 100 °C, (d) H2O 120 °C, (e) EtOH 120 °C, (f) DMF 150 °C, (g) DMF 200 °C for 24 h. The figure under the SEM image shows the corresponding statistical distribution of fiber diameters and its Gaussian fit. (h) Optical image of sample N-3 immersed in DMF at 200 °C for 24 h. | |
Atomic force microscopy (AFM) as one of the foremost tools for reflecting and measuring the surface morphology of the matter at the nanoscale has also been used to analyse the nanofibers. Fig. 9 presents the AFM 3D and 2D images revealing the changes in the topographical structure of the prepared nanofibers before and after the immersion. Fig. 9a shows the surface morphology of two paralleled fibers together. After the immersion experiment, the morphology of the nanofibers kept intact but the surface became rougher slightly. The image profile (Fig. S7†) also show that the surface height fluctuation increased from 10 nm to 20–30 nm.
 |
| | Fig. 9 AFM images of (a) sample N-3: two paralleled fibers, (b) sample N-3 immersed in DMF 200 °C for 24 h: a single fiber. | |
Mechanical properties
The mechanical properties of the nanofibers are shown in Table 2. The polymer nanofibers (sample N-0) exhibit great tensile strength, poor breaking elongation and a yield strength of zero. These figures are in accordance with the fact that sample N-0 became rigid after the thermal treatment. Adding silica content to the nanofibers reduced the tensile strength significantly from 7.26 to 0.77 MPa (sample N-1). On the other hand, the breaking elongation of the nanofibers increased from 7.03 to 16.72% and the yield strength increased to 0.11 MPa. With increasing silica content, the tensile strength, breaking elongation and yield strength all rose to some extent. Although the tensile strength of sample N-4 was still much lower than that of sample N-0, the breaking elongation of the composite nanofibers was almost eight times larger than the polymer nanofibers. These results indicate that the composite nanofibers are malleable and can sustain some strength at yield.42 This explains that the composite nanofibers possess remarkable solvent and temperature resistance and the underlined reason is the silica content forms a network structure with the two polymers via chemical bonding which enhances the stability of the whole materials.
Table 2 Mechanical properties of the ternary nanofibers
| Sample No. |
Tensile strength (MPa) |
Breaking elongation (%) |
Yield strength (MPa) |
| N-0 |
7.26 |
7.03 |
0 |
| N-1 |
0.77 |
16.72 |
0.11 |
| N-2 |
0.91 |
25.95 |
0.13 |
| N-3 |
1.51 |
45.57 |
0.26 |
| N-4 |
2.31 |
60.47 |
0.33 |
Conclusions
In summary, a novel organic–inorganic ternary nanofibers were successfully produced by cooperative fabrication. Through adjusting the mass ratio of the silica gel and polymer gel and the electrospinning parameters, straight and homogeneous nanofibers were obtained. This PVA/PAA/SiO2 composite nanofibers exhibited remarkable stability in eight common solvents at various temperatures. After immersing in these solvents for 48 h, little changes in the morphology and diameter distribution of the fibers were found. In the aspect of temperature, the fibers remain stable under the boiling point of all solvents. For DMF the nanofibers still kept intact even the temperature was raised up to 200 °C. The ternary electrospun nanofibers also showed great mechanical properties especially good at malleability with a high breaking elongation. The remarkable solvent and temperature resistance of the composite nanofibers as well as the great mechanical property might make the material applicable in some areas which are not practical before. Future researches would be focused on the functionalization of the fibers to improve the properties and extend its application fields.
Acknowledgements
This study is part of the Africa – China Cooperation Program on Environment which is supported by the Ministry of Science and Technology of China (MOST) and coordinated by the United Nations Environment Programme (no. 2010DFA92820 and no. 2010DFA92800). We also acknowledge the financial support from the National Science Foundation of China (51203117).
Notes and references
- L. Zhang, J. Luo, T. J. Menkhaus, H. Varadaraju, Y. Sun and H. Fong, J. Membr. Sci., 2011, 369, 499–505 CrossRef CAS PubMed.
- T. Subbiah, G. Bhat, R. Tock, S. Parameswaran and S. Ramkumar, J. Appl. Polym. Sci., 2005, 96, 557–569 CrossRef CAS.
- Z.-M. Huang, Y. Z. Zhang, M. Kotaki and S. Ramakrishna, Compos. Sci. Technol., 2003, 63, 2223–2253 CrossRef CAS.
- J. D. Schiffman and C. L. Schauer, Polym. Rev., 2008, 48, 317–352 CrossRef CAS.
- S. Kalluri, K. H. Seng, Z. Guo, H. K. Liu and S. X. Dou, RSC Adv., 2013, 3, 25576–25601 RSC.
- I. S. Chronakis, J. Mater. Process. Technol., 2005, 167, 283–293 CrossRef CAS PubMed.
- Z. Sun, J. M. Deitzel, J. Knopf, X. Chen and J. W. Gillespie, J. Appl. Polym. Sci., 2012, 125, 2585–2594 CrossRef CAS.
- L. Jia, M. P. Prabhakaran, X. Qin, D. Kai and S. Ramakrishna, J. Mater. Sci., 2013, 1–12 Search PubMed.
- L. Huang, S. S. Manickam and J. R. McCutcheon, J. Membr. Sci., 2013, 436, 213–220 CrossRef CAS PubMed.
- Y.-E. Miao, S. He, Y. Zhong, Z. Yang, W. W. Tjiu and T. Liu, Electrochim. Acta, 2013, 99, 117–123 CrossRef CAS PubMed.
- T. A. Arun, A. A. Madhavan, D. K. Chacko, G. S. Anjusree, T. G. Deepak, S. Thomas, S. V. Nair and A. S. Nair, Dalton Trans., 2014, 43, 4830–4837 RSC.
- S. Wu, F. Li, Y. Wu, R. Xu and G. Li, Chem. Commun., 2010, 46, 1694–1696 RSC.
- E. Baştürk, S. Demir, Ö. Danış and M. V. Kahraman, J. Appl. Polym. Sci., 2013, 127, 349–355 CrossRef.
- L. Li and Y.-L. Hsieh, Nanotechnology, 2005, 16, 2852 CrossRef CAS.
- Y. Liu, B. Bolger, P. A. Cahill and G. B. McGuinness, Mater. Lett., 2009, 63, 419–421 CrossRef CAS PubMed.
- C. Shao, H.-Y. Kim, J. Gong, B. Ding, D.-R. Lee and S.-J. Park, Mater. Lett., 2003, 57, 1579–1584 CrossRef CAS.
- G. D. Fu, L. Q. Xu, F. Yao, K. Zhang, X. F. Wang, M. F. Zhu and S. Z. Nie, ACS Appl. Mater. Interfaces, 2009, 1, 239–243 CAS.
- Y. Zhou, P. Qi, Z. Zhao, Q. Liu and Z. Li, RSC Adv., 2014, 4, 16731–16738 RSC.
- G. Poologasundarampillai, B. Yu, J. R. Jones and T. Kasuga, Soft Matter, 2011, 7, 10241 RSC.
- X. Liu, H. Zhang, Z. Tian, A. Sen and H. R. Allcock, Polym. Chem., 2012, 3, 2082 RSC.
- T. Pirzada, S. A. Arvidson, C. D. Saquing, S. S. Shah and S. A. Khan, Langmuir, 2012, 28, 5834–5844 CrossRef CAS PubMed.
- R. Xu, M. Jia, Y. Zhang and F. Li, Microporous Mesoporous
Mater., 2012, 149, 111–118 CrossRef CAS PubMed.
- Y. Xu, Y. Wen, Y.-n. Wu, C. Lin and G. Li, Mater. Lett., 2012, 78, 139–142 CrossRef CAS PubMed.
- D. Kim, H. Park, J. Rhim and Y. Lee, Solid State Ionics, 2005, 176, 117–126 CrossRef CAS PubMed.
- J.-P. Jeun, Y.-K. Jeon, Y.-C. Nho and P.-H. Kang, J. Ind. Eng. Chem., 2009, 15, 430–433 CrossRef CAS PubMed.
- S. Wu, F. Li, H. Wang, L. Fu, B. Zhang and G. Li, Polymer, 2010, 51, 6203–6211 CrossRef CAS PubMed.
- H. Yang, R. Xu, X. Xue, F. Li and G. Li, J. Hazard. Mater., 2008, 152, 690–698 CrossRef CAS PubMed.
- B. Kim, H. Park, S.-H. Lee and W. M. Sigmund, Mater. Lett., 2005, 59, 829–832 CrossRef CAS PubMed.
- L. Li and Y.-L. Hsieh, Polymer, 2005, 46, 5133–5139 CrossRef CAS PubMed.
- B. Ding, H.-Y. Kim, S.-C. Lee, C.-L. Shao, D.-R. Lee, S.-J. Park, G.-B. Kwag and K.-J. Choi, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 1261–1268 CrossRef CAS.
- S.-S. Choi, S. G. Lee, S. S. Im, S. H. Kim and Y. L. Joo, J. Mater. Sci. Lett., 2003, 22, 891–893 CrossRef CAS.
- L. Ji, A. J. Medford and X. Zhang, J. Polym. Sci., Part B: Polym. Phys., 2009, 47, 493–503 CrossRef CAS.
- F. Liu, Q. Q. Ni and Y. Murakami, Text. Res. J., 2013, 83, 510–518 Search PubMed.
- G. M. Wu, S. J. Lin and C. C. Yang, J. Membr. Sci., 2006, 275, 127–133 CrossRef CAS PubMed.
- W. L. Huang, S. H. Cui, K. M. Liang, Z. F. Yuan and S. R. Gu, J. Phys. Chem. Solids, 2002, 63, 645–650 CrossRef CAS.
- D. S. Kim, I. C. Park, H. I. Cho, D. H. Kim, G. Y. Moon, H. K. Lee and J. W. Rhim, J. Ind. Eng. Chem., 2009, 15, 265–269 CrossRef CAS PubMed.
- S. Xiao, M. Shen, H. Ma, R. Guo, M. Zhu, S. Wang and X. Shi, J. Appl. Polym. Sci., 2010, 116, 2409–2417 CAS.
- J. Zeng, H. Hou, J. H. Wendorff and A. Greiner, Polymer, 2004, 78, 1–8 Search PubMed.
- S. Xiao, M. Shen, R. Guo, S. Wang and X. Shi, J. Phys. Chem. C, 2009, 113, 18062–18068 CAS.
- E. Yang, X. Qin and S. Wang, Mater. Lett., 2008, 62, 3555–3557 CrossRef CAS PubMed.
- A. C. Patel, S. Li, C. Wang, W. Zhang and Y. Wei, Chem. Mater., 2007, 19, 1231–1238 CrossRef CAS.
- Z.-M. Huang, Y. Zhang, S. Ramakrishna and C. Lim, Polymer, 2004, 45, 5361–5368 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra03633f |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.