
Enhanced melt free radical grafting efficiency of polyethylene using a novel redox initiation method
Abstract
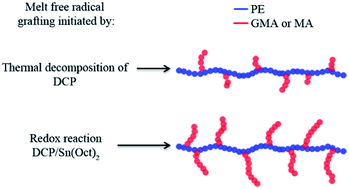
In this work, for the first time, redox initiation was employed in the melt free radical grafting of glycidyl methacrylate (GMA) and maleic anhydride (MA) onto polyethylene (PE) by reactive extrusion. Since it is very challenging to obtain high grafting degrees of GMA and MA onto polymer backbones through conventional free radical initiation, especially in the extrusion process, the strategy of using a peroxide/reducing agent initiator was introduced. The redox initiation system was composed of dicumyl peroxide (DCP) and Tin(II) 2-ethylhexanoate (Sn(Oct)2). The grafting reaction was monitored by torque rheometry and graft products were analyzed by Fourier Transform Infrared Spectroscopy. Effects of monomer concentration and DCP/Sn(Oct)2 ratio on the degree of grafting were studied. The redox initiation proved to be very effective in improving the grafting degree of GMA and MA onto PE. It is reasoned that, in the presence of Sn(Oct)2, the free radicals produced were not subject to a cage effect, yielding high initiator efficiency.
 Please wait while we load your content...
Please wait while we load your content...

