DOI:
10.1039/C4RA03569K
(Paper)
RSC Adv., 2014,
4, 25588-25595
Structural and functional studies on coordination polymers based on 5-tert-butylisophthalic acid and N,N′-bis-(4-pyridylmethyl) piperazine†
Received
19th April 2014
, Accepted 2nd June 2014
First published on 2nd June 2014
Abstract
By using 5-tert-butylisophthalic acid (H2tbip) and N,N′-bis-(4-pyridylmethyl) piperazine (bpmp), three coordination polymers formulated as {[Co(bpmp)(tbip)]·H2O}n (1), {[Ni(bpmp)1.5(tbip)(H2O)]·H2O}n (2), and {[Cd(bpmp)1.5(tbip)]·3H2O}n (3) have been synthesized under hydrothermal conditions. All complexes were characterized by single crystal X-ray diffraction analysis, powder X-ray diffraction analysis, elemental analysis, infrared spectroscopy and thermogravimetric analysis. The results show that complex 1 presents a 3D 6-connected network. Complex 2 shows a three-fold interpenetrating framework based on subunits with {46.64}-bnn hexagonal BN topology while complex 3 shows a 2D → 3D polycatenation framework containing 2D bilayer motifs as the fundamental building units. The photocatalytic properties of complexes 1 and 2 have been evaluated and the results show that they are active as a catalyst for the decomposition of Rhodamine B under visible light. The photoluminescence properties of complex 3 were also assessed in the solid state at room temperature.
Introduction
Over the past three decades, the design and construction of metal–organic coordination polymers (MOCPs) have received great attention from chemists not only due to their intriguing variety of architectures and topologies,1 but also because of their potential applications in areas such as gas storage, ion exchange, sensors or catalysis.2 From the point of view of crystal engineering, the synthetic strategies to target new hybrid complexes typically emphasize the use of the specific local coordination geometry of the central metal cation, as well as the configuration and coordination geometry and size of the ligand, to help control the topological structure and potential property of the product that final forms under certain reaction condition.3 By following this approach, the structure and desired property of MOCPs can be modulated to some degree. But precise control of the structure and property of MOCPs is still difficult to achieve because many factors such as temperature, pH value, counterion, solvent play important role on the construction process. Therefore, it is undoubtedly important for further investigation of how these factors have effect on the assemble process.
In the context of developing MOCPs with desired property, several approaches have been developed including incorporation of active metal ions or cluster centers into the frameworks,4 postsynthetic modification of MOCPs,5 custom-designing of the functional organic ligands such as inclusion of substituent moieties on the organic ligand.6 For example, by using amino–terephthalic acid Couck and co-authors reported the well-known MIL-53 with large separation power for CO2 and CH4.6a We have long been focusing on the construction of MOCPs with interesting architectures and potential applications based on flexible N-coordinated pyridyl ligand and substituent aromatic acids.7 For example, we have successfully synthesized several compounds with interesting 2D → 3D polycatenation or interpenating networks based on N,N′-bis-(4-pyridyl-methyl) piperazine (bpmp) ligand and 5-methylisophthalic acid ligand in our previous studies.7a A series of coordination polymers based on bpmp ligand have also been reported by LaDuca and co-workers.8 These results indicate that bpmp ligand is effective tecton in the construction of coordination complexes with interesting structures. Bearing the aforementioned ideas in mind, we now expand our work to bpmp ligand and rigid 5-tert-butylisophthalic acid ligand system. The above mentioned mixed ligand system was chosen to build entangled MOCPs owing to the following two considerations: (a) it is well known that mixed ligand systems provide more coordination modes and increase structure diversity of the outcome compounds.9 Thus, they are required to realize entangled networks with interesting topological structures. (b) The substituent isobutyl group in the 5-tert-butylisophthalic acid ligand acting as auxochromic and bathochromic group may shift the absorption wavelength and promote charge transfer interactions of the resulting compounds, which is promising for the development of luminescent probes and visible-light photocatalysts. (c) The 5-tert-butylisophthalic acid ligand with two carboxyl groups and a isobutyl group has been somewhat seldom involved in the assemble of MOCPs. Noticeably, one feature of the H2tbip ligand is that it is anionic when coordinating to metal cation and thus fulfills the charge balance of the coordination complex. This feature can prevent counter ions coordinating to the metal center, which usually happens in a neutral ligand system and reduce the coordination variety between metal cation and the ligand. Now, this time we successfully synthesize three coordination architectures with interesting topological structures based on mixed bpmp and the 5-tert-butylisophthalic acid ligand system. In the present work, we give a report on the synthesis and characterization of three coordination compounds formulated as {[Co(bpmp)(tbip)]·H2O}n (1), {[Ni(bpmp)1.5(tbip)(H2O)]·H2O}n (2) and {[Cd(bpmp)1.5(tbip)]·3H2O}n (3). Their structures were determined by single-crystal X-ray diffraction analyses and further characterized by elemental analysis, infrared spectrum (IR), thermogravimetric analysis (TGA) and powder X-ray diffraction (PXRD). In addition, photocatalytic properties of complexes 1 and 2 for the decomposition of Rhodamine B under visible light and luminescent property of compound 3 has also been explored and discussed in detail.
Experimental details
Materials and general methods
All commercially available reagents and starting materials were of reagent-grade quality and used without further purification. A piperazine–pyridine ligand, 1,4-bis-(4-pyridyl-methyl) piperazine (bpmp), was prepared according to a previously reported procedure.10 Elemental analyses (C, H, N) were carried out on an Elementar Vario EL III analyzer. Infrared (IR) spectra were recorded on PerkinElmer Spectrum One as KBr pellets in the range 4000–400 cm−1. Thermogravimetric analysis was recorded with a Perkin-Elmer Diamond TG/DTA instrument at a heating rate of 10 °C min−1 under nitrogen atmosphere. X-ray powered diffraction (XRPD) patterns of the samples were recorded by an X-ray diffractometer (Rigaku D/Max 2200PC) with a graphite monochromator and CuKα radiation at room temperature while the voltage and electric current are held at 40 kV and 20 mA. UV-visible absorption spectra were collected in Hitachi U-4100. Solid-state emission and excitation spectra were carried out on an Edinburgh FLS920 phosphorimeter equipped with a continuous Xe-900 xenon lamp and an nF900 ns flash lamp.
Synthesis of {[Co(bpmp)(tbip)]·H2O}n (1)
A mixture of Co(NO3)2·6H2O (29.1 mg, 0.1 mmol), bpmp (26.8 mg, 0.1 mmol) and H2tbip (22.2 mg, 0.10 mmol) was dispersed in mixed N,N-dimethylformamide (DMF) and deionized water (9 mL, v/v = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), and then sealed in a 25 mL Teflon-lined stainless steel autoclave, and heated at 150 °C for 72 hours, then slowly cooled to room temperature during 24 hours. Red block crystals were recovered by filtration, washed by distilled water, and dried in air at ambient temperature. Yield: 47% (based on Co). Calcd for C28H34N4O5Co (565.53): C 59.47, H 6.06, N 9.91; found: C 59.56, H 6.11, N 9.97. IR (KBr, cm−1): 3435 (s, br), 2950(w), 2815(m), 1618(s), 1534(m), 1458(m), 1371(s), 1161(m), 1014(m), 843(m), 726(m).
:1), and then sealed in a 25 mL Teflon-lined stainless steel autoclave, and heated at 150 °C for 72 hours, then slowly cooled to room temperature during 24 hours. Red block crystals were recovered by filtration, washed by distilled water, and dried in air at ambient temperature. Yield: 47% (based on Co). Calcd for C28H34N4O5Co (565.53): C 59.47, H 6.06, N 9.91; found: C 59.56, H 6.11, N 9.97. IR (KBr, cm−1): 3435 (s, br), 2950(w), 2815(m), 1618(s), 1534(m), 1458(m), 1371(s), 1161(m), 1014(m), 843(m), 726(m).
Synthesis of {[Ni(bpmp)1.5(tbip)(H2O)]·H2O}n (2)
A mixture of Ni(NO3)2·6H2O (29.1 mg, 0.1 mmol), bpmp (26.8 mg, 0.1 mmol), H2tbip (22.2 mg, 0.10 mmol) and NaOH (8.00 mg 0.2 mmol) in deionized water (6 mL), was sealed in a 25 mL Teflon-lined stainless steel autoclave, and heated at 130 °C for 72 hours, then slowly cooled to room temperature during 24 hours. Green block crystals were recovered by filtration, washed by distilled water, and dried in air at ambient temperature. Yield: 40% (based on Ni). Calcd for C36H46N6O6Ni (717.48): C 60.26, H 6.46, N 11.71; found: C 60.36, H 6.51, N 11.68. IR (KBr, cm−1): 3440 (s, br), 2943(w), 2815(m), 1619(s), 1537(m), 1427(m), 1371(s), 1295(m), 1012(m), 846(m), 784(w), 726(m), 496(w).
Synthesis of {[Cd(bpmp)1.5(tbip)]·3H2O}n (3)
Compound 3 was prepared in the same way as that for 2 but using Cd(NO3)2·6H2O (30.8 mg, 0.1 mmol). Colorless block crystals were recovered by filtration, washed by distilled water, and dried in air at ambient temperature. Yield: 44% (based on Cd). Calcd for C36H48N6O7Cd (789.21): C 54.79, H 6.13, N 10.65; found: C 54.87, H 6.18, N 10.73. IR (KBr, cm−1): 3440 (w, br), 2963(w), 2813(w), 1626(s), 1567(m), 1432(m), 1366(s), 1160(w), 1012(w), 838(w), 786(w), 734(w), 490(w).
Photodegradation experiments
Photocatalytic activity of complexes 1 and 2 was evaluated by photocatalytic degradation of RhB under visible light at ambient temperature. A tungsten filament lamp was used as a visible light source. In the typical experiment, 100 mL glass reactor was charged with 50 mL aqueous RhB solution (10 mg L−1) and 50 mg photocatalyst (1 or 2) was dispersed in it for 30 min in the absence of light to attain adsorption equilibrium. Subsequently, 2 mL of 30% M H2O2 solution was added and the pH value was adjusted to 3 with sulfuric acid (0.5 mol L−1). Then the reaction was illuminated by a tungsten filament lamp and aliquots of the reaction mixture were periodically taken and analyzed with a UV-vis spectrophotometer at an absorption wavelength of 552 nm. For comparison, the photodegradation process of RhB without any photocatalyst has also been studied under the same conditions.
Crystallographic details
Data collection were performed on an Xcalibur, Eos, Gemini diffractometer with graphite-monochromated Mo-Kα (λ = 0.71073 Å) radiation at room temperature. Data reduction is accomplished by the CrysAlisPro (Oxford Diffraction Ltd., version 1.171.33.55) program.11 Empirical absorption corrections were applied to the data using the SADABS program.12 The structures were solved by direct methods and refined by the full-matrix least-squares on F2 using the SHELXTL-97 program.13 All non-hydrogen atoms were refined with anisotropic displacement parameters. The positions of hydrogen atoms attached to carbon atoms were generated geometrically (C–H bond fixed at 0.99 Å). Idealized positions of H atoms belonging to nitrogen atoms and water molecules were located from Fourier difference maps and refined isotropically. Crystallographic data and structure determination summaries are listed in Table 1. Selected bond lengths and angles of complexes 1–3 are listed in Table S1.† The topological analysis and some diagrams were produced using TOPOS program.14
Table 1 Crystal data and structure refinement parameters for compoundsa
| Complex |
1 |
2 |
3 |
| R = Σ||Fo| − |Fc||/Σ|Fo|. wR(F2) = [Σw(Fo2 − Fc2)2/Σw(Fo2)2]1/2. |
| Empirical formula |
C28H34N4O5Co |
C36H46N6O6Ni |
C36H48N6O7Cd |
| Formula weight |
565.53 |
717.48 |
789.21 |
| Crystal system |
Triclinic |
Monoclinic |
Monoclinic |
| Space group |
P1 |
P21/c |
P21/c |
| a (Å) |
10.192(4) |
11.0054(3) |
17.0669(10) |
| b (Å) |
12.0704(19) |
17.5194(5) |
10.2827(7) |
| c (Å) |
12.0780(15) |
19.5049(5) |
21.854(2) |
| α (deg) |
91.254(12) |
90 |
90 |
| β (deg) |
98.01(3) |
105.175(3) |
95.664(5) |
| γ (deg) |
106.54(3) |
90 |
90 |
| V (Å3) |
1407.5(6) |
3629.57(17) |
3816.4(5) |
| Z |
2 |
4 |
4 |
| Dcalc. (g cm−3) |
1.330 |
1.306 |
1.363 |
| F(000) |
590 |
1504 |
1616 |
| Reflns collected/unique |
13760/5599 |
36444/7399 |
29564/8670 |
| μ (mm−1) |
0.653 |
0.587 |
0.626 |
| GOF on F2 |
1.056 |
1.058 |
1.057 |
| R1a(I > 2σ (I)) |
0.0453 |
0.0433 |
0.0513 |
| wR2b (all data) |
0.1231 |
0.1204 |
0.1423 |
Results and discussion
Description of structure 1
The single-crystal X-ray analysis reveals that complex 1 crystallizes in the triclinic P1 space group and exhibits a three-dimensional framework. As shown in Fig. 1a, the asymmetric unit of complex 1 contains one Co(II) cation, one fully deprotonated tbip2− anion, two haves of two crystallographically distinct bpmp units (marked by N1–N3 and marked by N2–N4, respectively) and one lattice water molecule. The local coordination geometry around the Co(II) center adopts a typical octahedron. It is coordinated by four oxygen atoms of two tbip2− anions and two nitrogen atoms of two bpmp molecules. The Co–O and Co–N bonds are in the range of 2.0858(19)–2.3327(18) Å and 2.137(2)–2.193(2) Å respectively (Table S1†), of which are similar to those reported for other Co(II) compounds.15 The tbip2− ligand adopts the η2μ2–η2 coordination mode with one of the two carboxylate groups displays a bidentate bridging coordination mode while the other one holds a bidentate chelating mode as shown in Scheme 1a. These tbip2− ligands link the Co(II) ions into 1D binuclear chains as shown in Fig. 1b. Further, each such chain is bridged by bpmp ligands to four adjacent identical motifs forming a 3D framework as shown in Fig. 1c. Topologically, each binuclear [Co2O4] cluster is connected to six other [Co2O4] clusters through four bpmp molecules and two tbip2− anions and is 6-connected. Both the bpmp molecule and two tbip2− anion are 2-connected. Therefore, complex 1 shows 3D 6-connected network with point symbol of {33.46.55.6} as shown in Fig. 1d. If the 2-connected nodes are transformed as links, the structure of 1 can be simplified as a 6-connected 3D net with the pcu topology.
 |
| | Fig. 1 (a) The coordination environment of Co(II) ion in 1; (b) view of the 1D chain of Co(II) linked by tbip2−; (c) view of the 3D framework in 1; (d) simplified 6-connected net in 1. Co, tbip2−, and bpmp ligands are simplified into red, blue, and green nodes, respectively. Hydrogen atoms and guest molecules are omitted for clarity. Symmetry codes: (A) 1 + x, 1 + y, 1 + z; (B) −x, 1 − y, 1 − z. | |
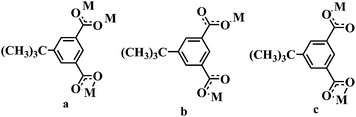 |
| | Scheme 1 Coordination modes of H2tbip in 1–3. | |
Description of structure 2
Single crystal X-ray diffraction analysis reveals that complex 2 crystallizes in the monoclinic space group P21/c and the asymmetric unit of 2 is constructed from one NiII ion, one fully deprotonated tbip2− ligand, one and a half bpmp ligands, one coordinated water molecule and one guest water molecule (Fig. 2a). The NiII ion is coordination by two carboxylic oxygen atoms from two tbip2− ligands, three pyridyl nitrogen atoms from three bpmp ligands and one oxygen atom from water molecule in a slightly distorted [NiO3N3] octahedral geometry. The two coordinated carboxyl O atoms together with two pyridyl N atoms define the equatorial positions, while the axial positions are occupied by one pyridyl N atom and the coordinated water molecule. The Ni–O bond distances are in the range of 2.0436(15)–2.1039(17) Å while the Ni–N bond distances are in the range of 2.0961(19)–2.1474(18) Å, which are all in good agreement with those reported for other nickel–oxygen and nickel–nitrogen donor compounds.16 The tbip2− ligand holds a η1–η1 coordination mode with each of the two carboxylate group bridging to one NiII ion as shown in Scheme 1b. The NiII centers are first linked by the tbip2− linkers to form 1D chain as shown in Fig. 2b. Meanwhile, the NiII centers are also bridged by bpmp ligands to form 2D layers with (6, 3) topology as shown in Fig. 2c. Then these chains and layers are cross-connected with each other to form five-connected 3D framework with {46.64}-bnn hexagonal BN topology in which the NiII ions act as uninodal center (Fig. 2d). Further investigation into the topology of complex 2 reveals that there are three identical independent 3D networks that interpenetrate with each other (Fig. 2e). The three 3D subunits are related to each other by one translation, placing the interpenetration into Class Ia (only one translation).17 In a word, the structure of 2 exhibits a 3D coordination network with three-fold interpenetration of subunits with {46.64}-bnn hexagonal BN topology.
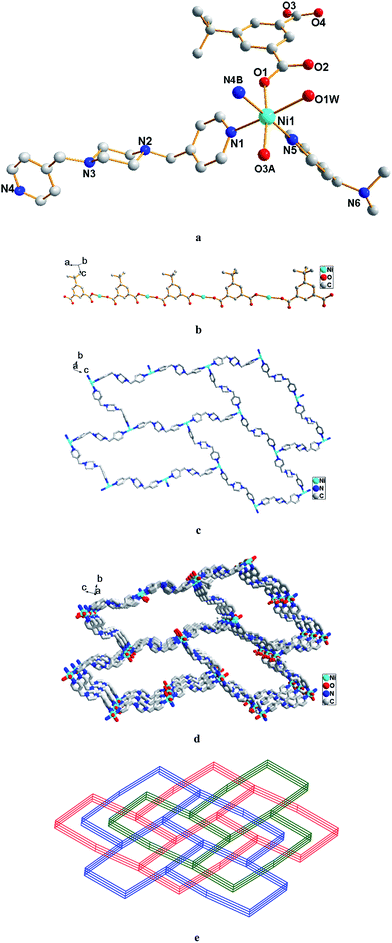 |
| | Fig. 2 (a) The coordination environment of Ni(II) ion in 2; (b) view of the 1D chain of Ni(II) linked by tbip2− in 2; (c) view of the (6, 3) layer of Ni(II) bridged by bpmp; (d) view of the 3D framework with {46.64}-bnn hexagonal BN topology; (e) schematic illustration of the three-fold interpenetration network in 2. Hydrogen atoms and guest molecules are omitted for clarity. Symmetry codes: (A) −1 + x, y, z; (B) 1 − x, 1/2 + y, −1/2 − z. | |
Description of structure 3
Determination of the structure of compound 3 by single crystal X-ray diffraction analysis indicates that compound 3 crystallizes in the monoclinic space group P21/c with one CdII ion, one fully deprotonated tbip2− ligand, one and a half bpmp ligands and three guest water molecules in the asymmetric unit (Fig. 3a). The CdII centers display distorted [CdN3O3] octahedral geometry with three carboxylic oxygen atoms from two tbip2− ligands and one pyridyl nitrogen atom from a bpmp ligand define the equatorial positions and two pyridyl nitrogen atoms from another two bpmp ligands occupies the axial positions. The tbip2− ligands in complexes 3 exhibit η2–η1 coordination mode with one of the two carboxylate groups adopts chelating coordination mode while the other one holds a monodentate bridging mode as shown in Scheme 1c. The coordination bond lengths that involve the Cd(II) center are in the range of 2.361(4)–2.416(4) Å for Cd–N bonds and 2.260(3)–2.571(3) Å for Cd–O bonds, which are all in good agreement with those typically observed (Table S1†).18 The CoII centers are first linked by the bpmp linkers in trans-configuration to form 1D ladder chain with (4,4) grids consist of cationic {[Cd(bpmp)]4}8+ squares with Cd⋯Cd distances of 17.0669(11) and 17.2073(15) Å. These square units are then joined by anionic tbip2− bridges to give two-dimensional bilayer motif with cuboidal box consists of eight Cd(II) atoms at the corners, showing large 1D channel (Fig. 3b). The large channel in the structure results in a fascinating and peculiar structural feature of 3, that is each layer motif is polycatenated with the two adjacent (the upper and the lower) identical motifs in a parallel fashion to give rise to a 3D polycatenated framework (Fig. 3c). The resulting 3D entanglement network is somewhat uncommon as only several examples of the species that comprised of two-level layers polycatenated with two adjacent ones have been reported.19
 |
| | Fig. 3 (a) The coordination environment of Cd(II) ion in 3; (b) view of the 2D bilayer subunit in 3; (c) schematic illustration of the polycatenation framework in 3. Hydrogen atoms and guest molecules are omitted for clarity. Symmetry codes: (A) x, 2 + y, 1 + z; (B) −1 + x, 1 + y, 1 + z. | |
Thermal analysis and PXRD results
Thermal stabilities of these new crystalline materials have been measured under nitrogen atmosphere to check the stability as shown in Fig. S2.† Complexes 1, 2 and 3 exhibit two main weight loss stages. In the TGA curve of complex 1, there are two continuous weight-loss steps. The first weight loss of 3.06% in the temperature range of 40 to 135 °C is equivalent of losing one lattice water molecule with a calculated value of 3.18%. The second weight-loss in the temperature from about 320 °C to 600 °C can be attributed to the collapse of the structure. For complex 2, a weight loss of about 4.93% occurs between 130 °C to 220 °C corresponds to the removal of the one coordinated water molecule and the one guest water molecule (calcd 5.02%). The second weight loss step start at about 220 °C and end at 560 °C can be ascribe to the collapse of the structure. For complex 3, the weight loss attributed to the release of the three uncoordinated water molecule is observed from 40 to 95 °C (obsd 6.80%, calcd 6.85%). The decomposition of the residual composition occurs from 285 to 560 °C. The final thermal decomposition products are unidentified because they are amorphous. The powder X-ray diffraction analyses of complexes 1–3 have also been carried out at room temperature. The experimental PXRD are in good agreement with the simulated patterns from the single-crystal structures as shown in Fig. S3,† which reveals the phase purity of the bulk crystalline materials.
UV-vis absorbance properties and photocatalytic activity
The solid-state UV-vis spectra of complexes 1–3 were measured at room temperature and the results are shown in Fig. S4.† The bands in the wavelength range 200–300 nm for complexes 1–3 could be assigned to the excitation of π-electrons of the aromatic system.20 Complex 1 and 2 show broad absorption bands in the visible light region (from about 450 nm to 550 nm for 1 and from 560 nm to 700 nm for 2), which are probably attributable to the d → d transitions of the central metal ions in the octahedron coordination field.21 For complex 3, no absorption were found in the visible light region due to the closed-shell electron configuration of d10 for Cd(II) ion.
Organic dyes such as MB and RhB are extensively used in industry fields and cause great environment pollution. Some coordination polymers have been reported showing photodegradation capacity for organic dyes under ultraviolet irradiation.22 Absorption of complexes 1 and 2 in visible light region indicated they may show absorption responses to visible light and may be used as heterogeneous photocatalysts for waste water treatment. Thus, the photocatalytic activity of as-prepared 1 and 2 was tested by the degradation of RhB solution under visible light irradiation. Analogous experiment was performed without 1 and 2 as described in the experimental section. The degradation experiment of RhB was tracked by visible spectroscopy and the results are depicted in Fig. 4. It was found that decomposition percentage of RhB within 6 h is about 76% in the presence of 1 and is about 69% in the presence of 2. For comparison, the photodegradation process of RhB without any photocatalyst has also been studied and the result suggests that about 12% RhB decomposition under the same condition (see also Fig. S5†). The above results clearly show that complexes 1 and 2 are effective catalysts for the decomposition of RhB under visible light in the presence of H2O2. The possible photocatalytic mechanism for the above degradation reactions can be explained based on some of the earlier observations.23 d → d transitions of the central metal ions in the octahedron coordination field occur upon visible light irradiation. Charge transfer promote electrons from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO). The electrons of the excited state in the LUMO are usually much activated and easily oxygenate H2O2 molecules to generate the ˙OH radicals which is known to have high activity to decompose the organic dyes.
 |
| | Fig. 4 Absorption spectra of a solution of RhB in the presence of compounds 1 and 2 under exposure to visible light (a and b); the photodegradation of RhB by compounds 1 and 2 monitored as the normalized change in concentration as a function of irradiation time (c). | |
Photoluminescent properties
Luminescent complexes are of great interest due to their potential application in chemical sensors, optical devices and various other fields.24 The solid state photoluminescent spectrum of complex 3 has been investigated at room temperature and the results are shown in Fig. 5. The emission spectrum has broad peaks with a maximum at 427 nm for complex 3 under excitation of 315 nm. There are typical two types of electronic excited state transition: ligand-to-ligand charge transfer (LLCT) and ligand-to-metal charge transfer (LMCT) in d10 metal coordination compounds.25 According to literature, the bpmp ligand show a fluorescent emission band at 386 nm under an excitation of 300 nm, while the free H2tbip ligand have emission band with a maximum of 345 nm with excitation upon 315 nm, both of which are probably attributable to the intra-ligand π* → π or π* → n transitions.21 Thus, the significant red-shifts of complex 3 compared to the emission spectra of the two free ligands may be tentatively ascribed to ligand-to-metal charge transfer (LMCT) that is in good agreement with literature examples on this class of coordination compounds.26
 |
| | Fig. 5 Solid-state fluorescence spectra of compound 3. | |
Conclusion
In summary, three new coordination complexes have been successfully synthesized under hydrothermal condition. Single crystal X-ray diffraction analyses show that complex 1 presents a 3D 6-connected network, complex 2 holds three-fold interpenetration framework based on subunits with {46.64}-bnn hexagonal BN topology while complex 3 presents 2D → 3D polycatenation framework containing 2D bilayer motifs as the fundamental building units. The photocatalytic activities of 1–2 were evaluated by the decomposition of RhB in aqueous solutions under visible light irradiation. Photoluminescent property of complex 3 has been studied in solid state at room temperature.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 21301069) and the Shandong Provincial Natural Science Foundation of China (no. ZR2012BQ004). C. Li, as a Taishan Scholar Endowed Professor, acknowledges the support from Shandong Province and UJN.
References
-
(a) J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213 RSC
 ;
(b) M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319 CrossRef CAS PubMed ;
(c) S. L. Qiu and G. S. Zhu, Coord. Chem. Rev., 2009, 253, 2891 CrossRef CAS PubMed ;
(d) W. J. Rieter, K. M. L. Taylor, H. Y. An and W. B. Lin, J. Am. Chem. Soc., 2006, 128, 9024 CrossRef CAS PubMed ;
(e) J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC ;
(f) H. Wu, J. Yang, Z. M. Su, S. R. Batten and J. F. Ma, J. Am. Chem. Soc., 2011, 133, 11406 CrossRef CAS PubMed ;
(g) S. R. Batten, Metal-Organic Frameworks, John Wiley & Sons, Inc., 2010 Search PubMed .
;
(b) M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319 CrossRef CAS PubMed ;
(c) S. L. Qiu and G. S. Zhu, Coord. Chem. Rev., 2009, 253, 2891 CrossRef CAS PubMed ;
(d) W. J. Rieter, K. M. L. Taylor, H. Y. An and W. B. Lin, J. Am. Chem. Soc., 2006, 128, 9024 CrossRef CAS PubMed ;
(e) J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC ;
(f) H. Wu, J. Yang, Z. M. Su, S. R. Batten and J. F. Ma, J. Am. Chem. Soc., 2011, 133, 11406 CrossRef CAS PubMed ;
(g) S. R. Batten, Metal-Organic Frameworks, John Wiley & Sons, Inc., 2010 Search PubMed . -
(a) Y. Sakata, S. Furukawa, M. Kondo, K. Hirai, N. Horike, Y. Takashima, H. Uehara, N. Louvain, M. Meilikhov, T. Tsuruoka, S. Isoda, W. Kosaka, O. Sakata and S. Kitagawa, Science, 2013, 339, 193 CrossRef CAS PubMed ;
(b) H. Wu, Q. Gong, D. H. Olson and J. Li, Chem. Rev., 2012, 112, 836 CrossRef CAS PubMed ;
(c) P. Y. Wu, C. He, J. Wang, X. J. Peng, X. Z. Li, Y. L. An and C. Y. Duan, J. Am. Chem. Soc., 2012, 134, 14991 CrossRef CAS PubMed ;
(d) J. Xiao, L. Qiu, F. Ke, Y. Yuan, G. Xu, Y. Wang and X. Jiang, J. Mater. Chem. A, 2013, 1, 8745 RSC ;
(e) J. H. Wang, M. Li and D. Li, Chem. Sci., 2013, 4, 1793 RSC ;
(f) X. L. Zhao, H. Y. He, T. H. Hu, F. N. Dai and D. F. Sun, Inorg. Chem., 2009, 48, 8057 CrossRef CAS PubMed .
-
(a) R. Haldar and T. K. Maji, CrystEngComm, 2013, 15, 9276 RSC ;
(b) S. Kitagawa and R. Matsuda, Coord. Chem. Rev., 2007, 251, 2490 CrossRef CAS PubMed ;
(c) N. J. Young and B. P. Hay, Chem. Commun., 2013, 49, 1354 RSC ;
(d) M. G. Goesten, F. Kapteijn and J. Gascon, CrystEngComm, 2013, 15, 9249 RSC .
-
(a) S. S. Mondal, A. Bhunia, A. Kelling, U. Schilde, C. Janiak and H. J. Holdt, J. Am. Chem. Soc., 2014, 136, 44 CrossRef CAS PubMed ;
(b) D. X. Ren, N. Xing, H. Shan, C. Chen, Z. Y. Cao and Y. H. Xing, Dalton Trans., 2013, 42, 5379 RSC ;
(c) Q. H. Chen, F. L. Jiang, D. Q. Yuan, G. X. Lyu and M. C. Hong, Chem. Sci., 2014, 5, 483 RSC ;
(d) Y. L. Hou, R. W. Y. Sun, X. P. Zhou, J. H. Wang and D. Li, Chem. Commun., 2014, 50, 2295 RSC .
-
(a) M. Kim, J. F. Cahill, H. H. Fei, K. A. Prather and S. M. Cohen, J. Am. Chem. Soc., 2012, 134, 18082 CrossRef CAS PubMed ;
(b) X. S. Wang, M. Chrzanowski, L. Wojtas, Y. S. Chen and S. Q. Ma, Chem.–Eur. J., 2013, 19, 3297 CrossRef CAS PubMed ;
(c) F. Sun, Z. Yin, Q. Q. Wang, D. Sun, M. H. Zeng and M. Kurmoo, Angew. Chem., Int. Ed., 2013, 52, 4538 CrossRef CAS PubMed ;
(d) G. Q. Kong, S. Ou, C. Zou and C. D. Wu, J. Am. Chem. Soc., 2012, 134, 19851 CrossRef CAS PubMed .
-
(a) K. K. Bisht, Y. Rachuri, B. Parmar and E. Suresh, RSC Adv., 2014, 4, 7352 RSC ;
(b) L. L. Wen, L. Zhou, B. G. Zhang, X. G. Meng, H. Qu and D. F. Li, J. Mater. Chem., 2012, 22, 22603 RSC ;
(c) R. B. Ferreira, P. M. Scheetz and A. L. B. Formiga, RSC Adv., 2013, 3, 10181 RSC .
-
(a) B. Xu, X. Lin, Z. Z. He, Z. J. Lin and R. Cao, Chem. Commun., 2011, 47, 3766 RSC ;
(b) B. Xu, J. Lü and R. Cao, Cryst. Growth Des., 2009, 9, 3003 CrossRef CAS ;
(c) B. Xu, Z. J. Lin, L. W. Han and R. Cao, CrystEngComm, 2011, 13, 440 RSC ;
(d) B. Xu, G. L. Li, H. X. Yang, S. Y. Gao and R. Cao, Inorg. Chem. Commun., 2011, 14, 493 CrossRef CAS PubMed .
-
(a) M. P. Martin, M. R. Montney, R. M. Supkowski and R. L. LaDuca, Cryst. Growth Des., 2008, 8, 3091 CrossRef ;
(b) K. M. Blake, G. A. Farnum, L. L. Johnston and R. L. LaDuca, Inorg. Chim. Acta, 2010, 363, 88 CrossRef CAS PubMed ;
(c) G. A. Farnum and R. L. LaDuca, Cryst. Growth Des., 2010, 10, 1897 CrossRef CAS ;
(d) D. P. Richard, R. J. Staples and R. L. LaDuca, Inorg. Chem., 2008, 47, 9754 CrossRef PubMed .
-
(a) L. F. Ma, L. Y. Wang, M. Du and S. R. Batten, Inorg. Chem., 2010, 49, 365 CrossRef CAS PubMed ;
(b) H. Wang, F. Y. Yi, S. Dang, W. G. Tian and Z. M. Sun, Cryst. Growth Des., 2014, 14, 147 CrossRef CAS ;
(c) X. L. Wang, N. Han, H. Y. Lin, A. X. Tian, G. C. Li and J. W. Zhang, Dalton Trans., 2014, 43, 2052 RSC ;
(d) X. G. Guo, W. B. Yang, X. Y. Wu, Q. K. Zhang and C. Z. Lu, CrystEngComm, 2013, 15, 10107 RSC ;
(e) J. Z. Gu, A. M. Kirillov, J. Wu, D. Y. Lu, Y. Tang and J. C. Wu, CrystEngComm, 2013, 15, 10287 RSC .
- Y. Y. Niu, H. W. Hou, Y. L. Wei, Y. T. Fan, Y. Zhu and C. X. Du, Inorg. Chem. Commun., 2001, 4, 358 CrossRef CAS .
- Oxford Diffraction, CrysAlisPro, Version 1.171.33.55, Oxford Diffraction Ltd, Yarnton, Oxfordshire, 2010 Search PubMed .
- G. M. Sheldrick, SADABS Siemens Area Detector Absorption Correction Program, University of Göttingen, Göttingen, Germany, 1994 Search PubMed.
- G. M. Sheldrick, SHELXS-97, Program for Crystal Structure Solution and Refinement, University of Göttingen, 1997 Search PubMed .
- V. A. Blatov, IUCr, Comput. Comm. Newslett., 2006, 7, 4, see also http://www.topos.ssu.samara.ru.
-
(a) C. L. Zhang, M. D. Zhang, L. Qin and H. G. Zheng, Cryst. Growth Des., 2014, 14, 491 CrossRef CAS ;
(b) F. L. Liu, L. L. Zhang, R. M. Wang, J. Sun, J. Yang, Z. Chen, X. P. Wang and D. F. Sun, CrystEngComm, 2014, 16, 2917–2928 RSC .
-
(a) S. Sanda, S. Parshamoni, A. Adhikary and S. Konar, Cryst. Growth Des., 2013, 13, 5442 CrossRef CAS ;
(b) P. Samarasekere, X. Q. Wang, A. J. Jacobson, J. Tapp and A. Mller, Inorg. Chem., 2014, 53, 244 CrossRef CAS PubMed ;
(c) R. Patra, H. M. Titi and I. Goldberg, CrystEngComm, 2013, 15, 2863 RSC .
- V. A. Blatov, L. Carlucci, G. Ciani and D. M. Proserpio, CrystEngComm, 2004, 6, 378 RSC .
-
(a) H. Keypour, M. Shayesteh, M. Rezaeivala, F. Chalabian, Y. Elerman and O. Buyukgungor, J. Mol. Struct., 2013, 1032, 62 CrossRef CAS PubMed ;
(b) Y. Yang, P. Du, Y. Y. Liu and J. F. Ma, Cryst. Growth Des., 2013, 13, 4781 CrossRef CAS ;
(c) L. L. Zhang, Y. Guo, Y. H. Wei, J. Guo, X. P. Wang and D. F. Sun, J. Mol. Struct., 2013, 1038, 73 CrossRef CAS PubMed .
-
(a) I. A. Baburin, V. A. Blatov, L. Carlucci, G. Ciani and D. M. Proserpio, CrystEngComm, 2008, 10, 1822 RSC ;
(b) G. P. Yang, L. Hou, L. F. Ma and Y. Y. Wang, CrystEngComm, 2013, 15, 2561 RSC ;
(c) O. K. Farha, C. D. Malliakas, M. G. Kanatzidis and J. T. Hupp, J. Am. Chem. Soc., 2010, 132, 950 CrossRef CAS PubMed .
-
(a) M. K. Nazeeraddin, S. M. Zakeeruddin and K. Kalyanasundaran, J. Phys. Chem., 1993, 97, 9607 CrossRef ;
(b) H. Khajavi, J. Gascon, J. M. Schins, L. D. A. Siebbeles and F. Kapteijn, J. Phys. Chem. C, 2011, 115, 12487 CrossRef CAS .
-
(a) H. X. Yang, T. F. Liu, M. N. Cao, H. F. Li, S. Y. Gao and R. Cao, Chem. Commun., 2010, 46, 2429 RSC ;
(b) K. K. Bisht and E. Suresh, Inorg. Chem., 2012, 51, 9577 CrossRef CAS PubMed .
-
(a) P. Mahata, G. Madras and S. Natarajan, J. Phys. Chem. B, 2006, 110, 13759 CrossRef CAS PubMed ;
(b) D. X. Li, C. Y. Ni, M. M. Chen, M. Dai, W. H. Zhang, W. Y. Yan, H. X. Qi, Z. G. Ren and J. P. Lang, CrystEngComm, 2014, 16, 2158 RSC ;
(c) D. Marcinkowski, M. Chorab, V. Patroniak, M. Kubicki, G. Kadziotka and B. Michalkiewicz, New J. Chem., 2014, 38, 604 RSC .
-
(a) Z. T. Yu, Z. L. Liao, Y. S. Jiang, G. H. Li and J. S. Chen, Chem.–Eur. J., 2005, 11, 2642 CrossRef CAS PubMed ;
(b) M. C. Das, H. Xu, Z. Y. Wang, G. Srinivas, W. Zhou, Y. F. Yue, V. N. Nesterov, G. D. Qian and B. L. Chen, Chem.
Commun., 2011, 47, 11715 CAS ;
(c) F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bachmann, Advanced Inorganic Chemistry, John Wiley & Sons, Inc., New York, 1999 Search PubMed .
-
(a) K. Jayaramulu, R. P. Narayanan, S. J. George and T. K. Maji, Inorg. Chem., 2012, 51, 10089 CrossRef CAS PubMed ;
(b) M. P. Suh, Y. E. Cheon and E. Y. Lee, Coord. Chem. Rev., 2008, 252, 1007 CrossRef CAS PubMed ;
(c) J. W. Zhang, H. T. Zhang, Z. Y. Du, X. Q. Wang, S. H. Yu and H. L. Zhang, Chem. Commun., 2014, 50, 1092 RSC ;
(d) Y. A. Li, S. K. Ren, Q. K. Liu, J. P. Ma, X. Y. Chen, H. M. Zhu and Y. B. Dong, Inorg. Chem., 2012, 51, 9629 CrossRef CAS PubMed ;
(e) X. Y. Zhou, P. X. Li, Z. H. Shi, X. L. Tang, C. Y. Chen and W. S. Liu, Inorg. Chem., 2012, 51, 9226 CrossRef CAS PubMed .
-
(a) M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330 RSC ;
(b) W. G. Lu, L. Jiang, X. L. Feng and T. B. Lu, Cryst. Growth Des., 2006, 6, 564 CrossRef CAS ;
(c) Y. X. Zhang, J. Yang, W. Q. Kan and J. F. Ma, CrystEngComm, 2012, 14, 6004 RSC ;
(d) L. L. Wen, Y. Z. Li, Z. D. Lu, J. G. Lin, C. Y. Duan and Q. J. Meng, Cryst. Growth Des., 2006, 6, 530 CrossRef CAS .
-
(a) J. Liu, H. B. Zhang, Y. X. Tan, F. Wang, Y. Kang and J. Zhang, Inorg. Chem., 2014, 53, 1500 CrossRef CAS PubMed ;
(b) Q. B. Bo, H. Y. Wang and D. Q. Wang, New J. Chem., 2013, 37, 380 RSC .
Footnote |
| † Electronic supplementary information (ESI) available: Powder XRD patterns, IR spectra, UV spectra and TG curves and table of collected bonds and angles. CCDC 993066, 981096 and 981097. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra03569k |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 




