
Pressure-sensitive adhesives based on soybean fatty acids†
Abstract
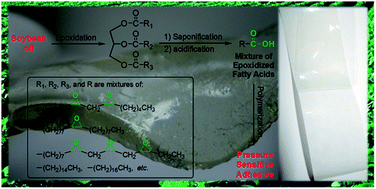
A new class of renewable pressure-sensitive adhesive (PSA) designed and developed from soybean oil was reported in this study. Soybean oil was epoxidized and hydrolysed selectively on the ester groups to afford a mixture of epoxidized fatty acids (EFAs) which were characterized by FTIR and 1H NMR spectroscopy. The EFA mixture without further purification was then polymerized directly in the presence and absence of a small amount of dicarboxylic acid compounds to afford hydroxyl-functionalized polymers. The peel strength, loop tack, shear strength and viscoelastic properties of the resulting (co)polymers revealed that the (co)polymers were suitable for PSA applications. The new PSAs could be fully bio-based and potentially biodegradable, and their preparation and application did not require the use of an organic solvent or a toxic chemical, thus being environmentally friendly.
 Please wait while we load your content...
Please wait while we load your content...

