
Detection of the neurotransmitter dopamine by a glassy carbon electrode modified with self-assembled perovskite LaFeO3 microspheres made up of nanospheres
Abstract
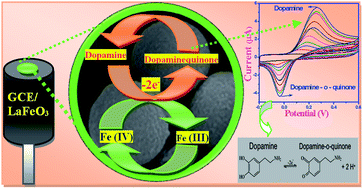
In this paper we report the detection of the neurotransmitter dopamine by an LaFeO3 microsphere-modified electrode in which the microspheres are made up of nanospheres. The morphology, structure and composition of the prepared nanostructure were characterized using SEM, TEM, XRD and XPS, and the electrocatalytic properties were investigated using cyclic voltammetry and amperometric studies. The modified electrodes markedly increased the efficiency of the electrocatalytic oxidation of dopamine. The biosensor exhibited high sensitivity at a low detection limit of 59 nM and wide linear range from 2 × 10−8 to 1.6 × 10−6 M (R = 0.9983). More importantly, the sensor effectively avoids the interference of ascorbic acid and uric acid. A possible electrocatalytic mechanism has been proposed. The LaFeO3 microspheres are highly promising for the detection of dopamine because of their high selectivity, fast response and good sensitivity.
 Please wait while we load your content...
Please wait while we load your content...

