The effect of alkylation, protonation, and hydroxyl group substitution on reversible alcohol and water addition to 2- and 4-formyl pyridine derivatives
Sanmitra Barman,
Katharine L. Diehl and
Eric V. Anslyn*
Department of Chemistry, The University of Texas, Austin, TX 78712, USA. E-mail: anslyn@austin.utexas.edu
First published on 11th June 2014
Abstract
The formation of acetals and hemiacetals from carbonyl species and alcohols is a well-studied reaction. For purposes of reversible covalent bonding, a thermodynamically favored reaction that is also kinetically fast is desirable. Toward this goal, the regiochemical influence of hydroxyl or methoxy substitution on 2- and 4-formyl pyridine derivatives on alcohol and water addition was studied. The goal was to investigate carbonyl activation via the electron deficient pyridine ring and the roles of intramolecular general acid catalysis/hydrogen bonding catalysis from an adjacent hydroxyl and of resonance donation from an adjacent hydroxyl or methoxy group in these addition reactions. A qualitative screen of formyl pyridine derivatives at room temperature in neutral and acidic conditions for the formation of addition products was undertaken. In subsequent studies, N-alkylated formyl pyridine derivatives were used in order to take advantage of the activation of the carbonyl provided by the positively charged pyridine ring under neutral conditions. The presence of a hydroxyl group adjacent to the aldehyde generally deactivates it to alcohol or water addition, particularly under neutral conditions when the hydroxyl group is deprotonated. Based on our findings, resonance donation rather than intramolecular general acid catalysis/hydrogen bonding catalysis appears to be the dominant effect behind this deactivation for the pyridinium derivatives. However, the presence of a hydroxyl or methoxy group stabilizes the oxocarbenium ion intermediate that results in acetal formation and thus is essential for forming acetal under neutral conditions.
Introduction
Hydroxyl groups are among the most prevalent of organic functional groups, being present in many natural products such as terpenes, steroids, and saccharides.1 They are also a main component of complex mixtures such as wines and perfumes.2 Hence, they are attractive targets for molecular receptors.3 The most promising class of receptors for the recognition of alcohols are boronic acids which form reversible covalent bonds with cis-diols.4,5 The strength of the interaction of cis-diols with phenyl boronic acids depends on parameters such as sterics, the pKa of the boronic acid, and the pH of the media.6 Also, it is well known that the presence of an aminomethyl group at an ortho position of the phenyl boronic acid improves the thermodynamic favorability and the rate of cyclic boronate ester formation.7 We have proposed based on kinetic isotope effect studies that the ammonium group can act as an intramolecular general acid catalyst to speed the rate of alcohol exchange at the boronic acid.8 Such receptors are excellent for the recognition of cis-diols but show little activity for trans-diols.9 We propose an alternative recognition motif for alcohols that employs reversible covalent reactions with aldehydes and could exploit the weakly nucleophilic character of a hydroxyl group (Scheme 1). This approach could be used to recognize cis- and trans-diols with comparable ability. | ||
| Scheme 1 An analogy between o-aminomethyl phenylboronates (coming from the corresponding boronic acid) to an acetal (coming from an o-hydroxypyridyl aldehyde). | ||
Unfortunately, the slow rate of hemiacetal and acetal formation at room temperature with simple alkyl aldehydes and ketones makes such carbonyl structures unattractive as receptors.10 To facilitate alcohol addition, an electron withdrawing group such as trifluoromethyl has been placed adjacent to the carbonyl (eqn (1)).11 Recently, our group and the Lehn group independently reported the notion of chelating a metal adjacent to the carbonyl acceptor to facilitate alcohol addition (eqn (2)). Both groups reported that 2-acyl pyridines reversibly add secondary alcohols when activated by a Brønsted acid or by a Lewis acid such as zinc triflate (eqn (3)).12
 | (1) |
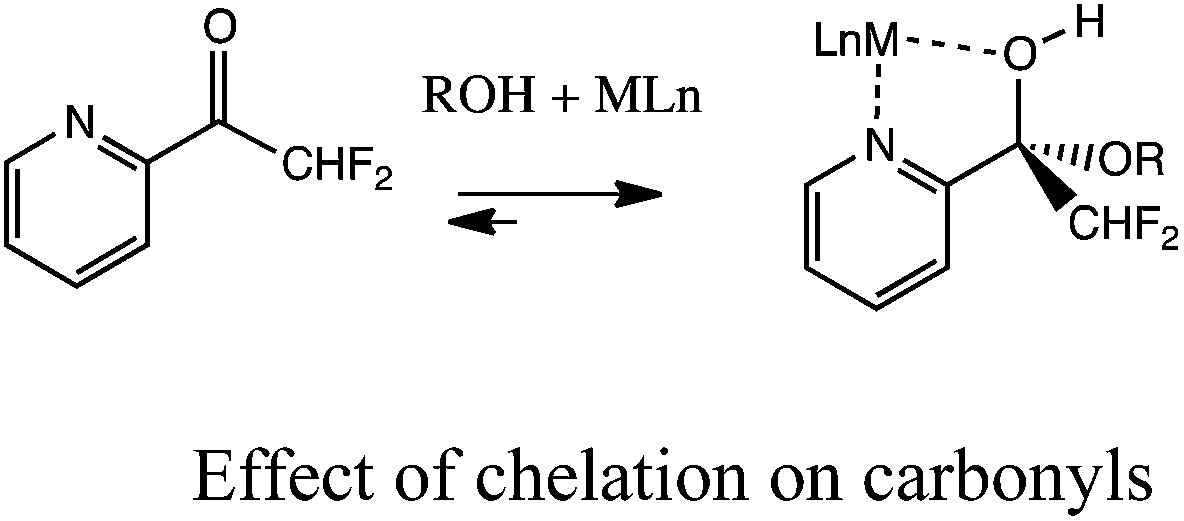 | (2) |
 | (3) |
 | (4) |
Most recently, we observed that placing a hydroxyl group ortho to the aldehyde carbonyl in 2-formyl pyridine enhanced the rate of condensation with benzyl amine in acetonitrile (eqn (4)) by a few orders of magnitude.13 Based on the preceding observations, we postulated that alcohols could analogously act as nucleophiles in a reaction such as the one shown in eqn (4) that is facilitated by the hydroxyl group and the pyridine ring.
The substrates used in this study are presented in Fig. 1. To test the role of the pyridine ring and the phenol, we report herein the reaction of various regioisomers of pyridine-based compounds containing a formyl and a hydroxyl group. Three roles of the hydroxyl group were postulated: (1) intramolecular general acid catalysis, (2) hydrogen bonding activation of the carbonyl group, or (3) resonance donation from the oxygen. To be an effective hydrogen bond donor or general acid catalyst, the hydroxyl group must be ortho to the aldehyde. Either effect is expected to enhance the rate of hemiacetal formation. While we likely cannot distinguish between the first two postulated roles for the hydroxyl group, we obtained derivatives in which the hydroxyl group was replaced by a methoxy group and in which the hydroxyl group was omitted altogether in order to study the third postulated role. By comparing the addition reactions to these derivatives, we can determine if the hydroxyl acts through hydrogen bonding or through resonance donation from the oxygen.
 | ||
| Fig. 1 Structures of compounds (1 to 7) used in this study. | ||
The first three compounds (1–3) have an aldehyde at the 2-position and a hydroxyl, methoxy, or hydrogen at the 3-position. These compounds were used to explore the electrostatic effects of the pyridine nitrogen in combination with the hydrogen bonding/general acid catalysis or resonance donation from an adjacent hydroxyl group to the carbonyl. The next three pyridine derivatives (4–6) have an aldehyde at the 4-position and a hydroxyl, methoxy, or hydrogen at the 3-position. These compounds were used to study differences in the electronic effects imposed by the pyridine nitrogen and the hydroxyl group based on the position of the aldehyde. Furthermore, the electron withdrawing nature of the pyridine nitrogen, which is enhanced by protonation or alkylation, was postulated to facilitate the addition of an alcohol to the carbonyl. This hypothesis was tested by including salicylaldehyde (7) in our studies. To follow the reactions, we used 1H-NMR spectroscopy.
A generalized mechanism of alcohol addition to an activated carbonyl with 2-formyl-3-hydroxypyridinium is shown in Scheme 2. The nucleophilic attack that leads to hemiacetal formation will be favored by an electron withdrawing pyridinium ring because the carbonyl carbon will be more electrophilic (A in Scheme 2). A hydroxyl adjacent to the aldehyde is expected to lower this electrophilicity due to the oxygen donor ability. However, the hydroxyl's hydrogen bond donor ability could enhance alcohol addition. In contrast, acetal formation should be favored by electron donating groups in the pyridine ring because acetal formation goes through an oxocarbenium ion intermediate (C in Scheme 2). Hence we expected that the presence of an electron donating hydroxyl group would have a mixed effect on the kinetics and thermodynamics of hemiacetal formation, whereas a hydroxyl or a methoxy group should both favor the formation of acetal.
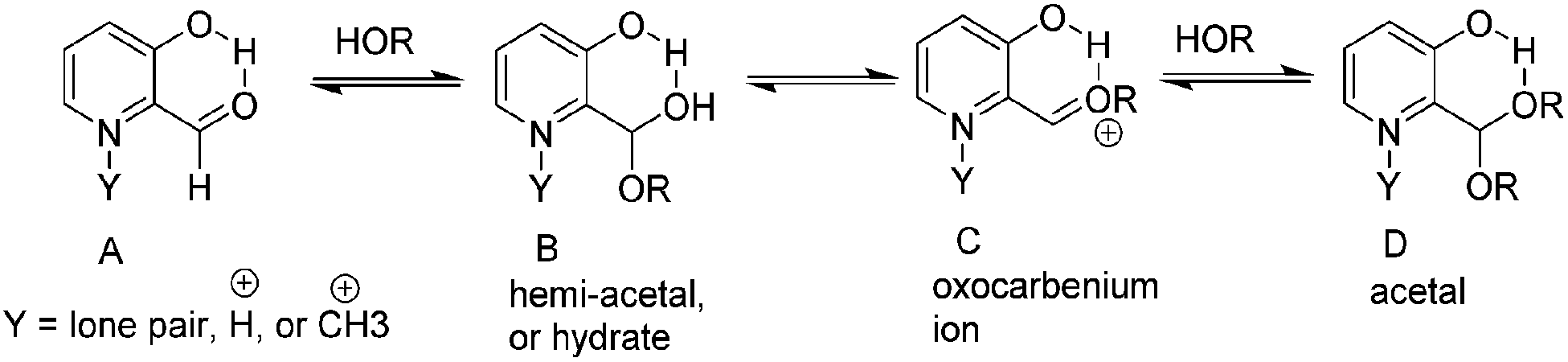 | ||
| Scheme 2 Addition of water or methanol to 2-formyl-3-hydroxypyridinium or N-alkylated derivative. | ||
Results and discussion
Design criteria
A series of 2- and 4-formyl pyridine derivatives (Fig. 1) were compared for hydrate formation in water, as well as hemiacetal and acetal formation in methanol at neutral and acidic pH. The potential for hydrogen bonding activation/intramolecular general acid catalysis was investigated as well by including 3-hydroxy and 3-methoxy derivatives of 2- and 4-formyl pyridine in the studies.In a second series of structures, the pyridine nitrogen was alkylated. The purpose of the alkylation was to introduce a positive charge on the nitrogen and hence to increase the electrophilicity of the carbonyl. By doing so, we sought to achieve similar rates and extents of reaction for these alkylated formyl pyridinium derivatives at neutral pH as was found for hemiacetal, acetal, and hydrate formation for the non-alkylated formyl pyridinium derivatives at acidic pH.
Hemiacetal, acetal, and hydrate formation from formyl pyridine derivatives
In order to explore the relative kinetics and thermodynamics of hemiacetal, acetal, and hydrate formation from the non-alkylated pyridine derivatives (compounds 1 to 7) in the absence and presence of acid, we carried out a series of alcohol and water addition experiments as described in Tables 1–4. Table 1 shows a comparison of hemiacetal and acetal formation with compounds 1–7 in methanol at neutral pH.
|
|||||||
|---|---|---|---|---|---|---|---|
| Compound | Aldehyde | Hemiacetal | Keq (He) | Acetal | Keq (Ac) | Time | |
| a In the tables, “instantaneous” indicates that the reaction occurred before the 1H-NMR could be taken. | |||||||
| 1 | A = N, X = CHO, Y = OH, Z = H | 50% | 41% | 0.82 | 9% | 0.18 | 7 days |
| 2 | A = N, X = CHO, Y = OCH3, Z = H | 18% | 82% | 4.55 | 0% | 0 | 2 days |
| 3 | A = N, X = CHO, Y = H, Z = H | 18% | 82% | 4.55 | 0% | 0 | 1 day |
| 4 | A = N, X = H, Y = OH, Z = CHO | 21% | 68% | 3.23 | 11% | 0.52 | 7 days |
| 5 | A = N, X = H, Y = OCH3, Z = CHO | 15% | 85% | 5.66 | 0% | 0 | Instantaneous |
| 6 | A = N, X = H, Y = H, Z = CHO | 13% | 87% | 6.69 | 0% | 0 | Instantaneous |
| 7 | A = C, X = CHO, Y = OH, Z = H | 100% | 0% | 0 | 0% | 0 | 7 days no reaction |
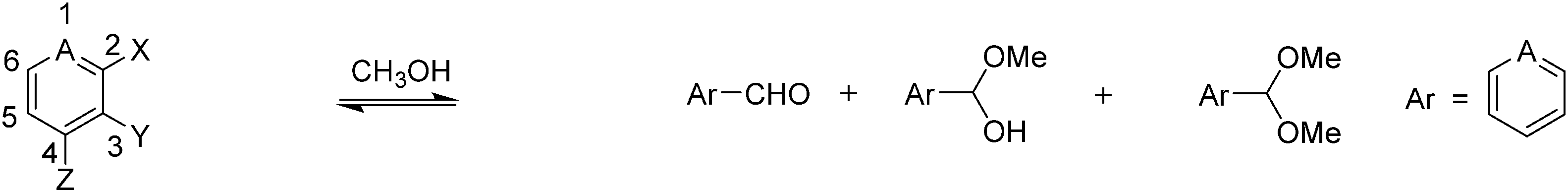
From the Hammet sigma values for hydroxy (−0.38) and methoxy groups (−0.27), one would expect to find that the derivatives with these groups behave similarly to one another but differently from the derivative with only a hydrogen if resonance donation was playing an important role.18 However, the results for compounds 2 and 3 were essentially identical to one another and were quite different from the results for compound 1. The pKa of the hydroxyl group in compound 1 has been measured to be about 6.8,19 so under neutral conditions about half of 1 in solution is in the deprotonated, anionic form. The deprotonated hydroxyl deactivates the carbonyl. Nevertheless, it is interesting that there is no measurable difference in the reactivity of 2 and 3, suggesting that the methoxy group essentially does not deactivate the aldehyde despite being an electron donating group.
For compound 1, some acetal was formed after seven days. However, for compounds 2 and 3, most of the aldehyde was converted to hemiacetal with no acetal formation. The implication of this finding is that hydrogen bonding is playing a role in the stabilization of the oxocarbenium ion intermediate (C in Scheme 2) that leads to acetal. While the hydroxyl anion deactivates the aldehyde to hemiacetal formation, it appears to be effective at stabilizing the oxocarbenium ion intermediate compared to a hydrogen or a methoxy group. Similar observations were obtained for compounds 5 and 6 compared to compound 4 in Table 1.
An interesting observation is that compounds 5 and 6 reached equilibrium faster than compounds 2 and 3 even though the product distributions for these reactions were ultimately the same at equilibrium. We postulate that the shorter reaction time for hemiacetal formation when the aldehyde is para to the pyridine nitrogen arises from steric interactions with solvent that is hydrogen bonded to the nitrogen that lowers the propensity to add solvent to the adjacent aldehyde. We observed no product formation with salicylaldehyde (7) after seven days. This result highlights the importance of the electron deficient pyridine ring for the activation of aldehydes toward alcohol addition under neutral conditions.
Table 2 shows the extent of hydrate formation at neutral pH for compounds 1–7. As with the hemiacetal/acetal formation experiments, compounds 2 and 3 appear to behave almost identically to one another. These results again demonstrate that the hydroxyl anion deactivates the carbonyl, while the methoxy group is not apparently deactivating. In the case of hydrate formation, compounds 2, 3, 5, and 6 all reach equilibrium immediately; however, a greater extent of aldehydes 5 and 6 (para) are converted to hydrate at equilibrium compared to aldehydes 2 and 3 (ortho). As described previously, this difference in the product distribution could be attributed to the steric influence imposed by the solvent molecules which are hydrogen bonded to the nitrogen of the pyridine ring.
|
|||||
|---|---|---|---|---|---|
| Compound | Aldehyde | Hydrate | Keq (Hy) | Time | |
| a In the tables, “instantaneous” indicates that the reaction occurred before the 1H-NMR could be taken. | |||||
| 1 | A = N, X = CHO, Y = OH, Z = H | 87% | 13% | 0.15 | 0.5 day |
| 2 | A = N, X = CHO, Y = OCH3, Z = H | 62% | 38% | 0.61 | Instantaneous |
| 3 | A = N, X = CHO, Y = H, Z = H | 60% | 40% | 0.66 | Instantaneous |
| 4 | A = N, X = H, Y = OH, Z = CHO | 68% | 32% | 0.47 | Instantaneous |
| 5 | A = N, X = H, Y = OCH3, Z = CHO | 44% | 56% | 1.27 | Instantaneous |
| 6 | A = N, X = H, Y = H, Z = CHO | 50% | 50% | 1.00 | Instantaneous |
| 7 | A = C, X = CHO, Y = OH, Z = H | 100% | 0% | 0 | 7 days no reaction |

Next we investigated the extent of hemiacetal and acetal formation from pyridinium derivatives, which are more electron deficient than the corresponding pyridine derivatives (Table 3). Hence, for compounds 1–5 addition of one equivalent of trifluoroacetic acid to the reaction results in more acetal formation compared to the corresponding neutral pyridine derivative reactions. Protonated compounds 1 and 2 are both converted entirely to acetal in about two days, while protonated compound 3 converts to about 50% acetal/50% hemiacetal upon reaching equilibrium after seven days. This finding implies that resonance donation is playing a role in the pyridinium reactions.
|
|||||||
|---|---|---|---|---|---|---|---|
| Compound | Aldehyde | Hemiacetal | Keq (He) | Acetal | Keq (Ac) | Time | |
| a In the tables, “instantaneous” indicates that the reaction occurred before the 1H-NMR could be taken. | |||||||
| 1 | A = N, X = CHO, Y = OH, Z = H | <1% | <1% | Almost 0 | 100% | >1000 | 1.5 days |
| 2 | A = N, X = CHO, Y = OCH3, Z = H | <1% | <1% | Almost 0 | 100% | >1000 | 2 days |
| 3 | A = N, X = CHO, Y = H, Z = H | 2% | 52% | 26 | 46% | 23 | 7 days |
| 4 | A = N, X = H, Y = OH, Z = CHO | <1% | 8% | >1000 | 92% | >1000 | 6 days |
| 5 | A = N, X = H, Y = OCH3, Z = CHO | 1% | 39% | 39 | 60% | 60 | 7 days |
| 6 | A = N, X = H, Y = H, Z = CHO | <1% | 100% | >1000 | <1% | 0 | Instantaneous |
| 7 | A = C, X = CHO, Y = OH, Z = H | 100% | <1% | Almost 0 | <1% | 0 | 7 days no reaction |

The reactions with the formyl group at the para position gave quite different results. In general, these reactions were slower and resulted in less acetal formation compared to their ortho substituted counterparts because the electron withdrawing effect from the positively charged nitrogen on the formyl group is stronger when the two groups are closer to one another. Protonated compound 4 is converted entirely to acetal within six days; while about 60% of protonated compound 5 is converted to acetal within seven days. In contrast, protonated compound 6 does not show any acetal formation. These results support the conclusion that resonance donation stabilizes the oxocarbenium ion (C in Scheme 2) in acetal formation, since the hydroxyl group is a better donor than the methoxy group, and the hydrogen has no donor ability. We can be confident that resonance donation from the methoxy group is occurring as evidenced by the greater formation of acetal from 2 compared to 3. It is also possible that the hydroxyl group is better at stabilizing the oxocarbenium intermediate than the methoxy because it can do so through hydrogen bond donor catalysis; however, it is not possible to conclude from this data which effect is responsible for the difference in acetal formation between the hydroxyl- (1) and methoxy-substituted (2) derivatives, and it is possible that both effects are occurring.
Lastly, we carried out experiments to screen the kinetics and thermodynamics of hydration for compounds 1 to 7 in acidic conditions (Table 4). All the reactions occurred instantaneously except for salicylaldehyde for which no reaction was observed after seven days. The extent of hydrate formation for these pyridinium derivatives was higher than their pyridine counterparts, which is again due to the increased electrophilicity of the aldehyde due to the electron withdrawing pyridinium nitrogen. The trend of the results for the hydroxyl-, methoxy-, and hydrogen-substituted 2- and 4-formyl pyridinium derivatives resembles those found for the corresponding pyridine derivatives. That is, the methoxy- and hydrogen-substituted derivatives behave essentially the same, and the hydroxyl-substituted derivatives result in somewhat less hydrate formation. This finding implies that hydrogen bonding is exerting an effect that somewhat inhibits the formation of hydrate.
|
|||||
|---|---|---|---|---|---|
| Compound | Aldehyde | Hydrate | Keq (Hy) | Time | |
| a In the tables, “instantaneous” indicates that the reaction occurred before the 1H-NMR could be taken. | |||||
| 1 | A = N, X = CHO, Y = OH, Z = H | 10% | 90% | 9 | Instantaneous |
| 2 | A = N, X = CHO, Y = OCH3, Z = H | 3% | 97% | 32.33 | Instantaneous |
| 3 | A = N, X = CHO, Y = H, Z = H | 0.5% | 99.5% | 199 | Instantaneous |
| 4 | A = N, X = H, Y = OH, Z = CHO | 25% | 75% | 3 | Instantaneous |
| 5 | A = N, X = H, Y = OCH3, Z = CHO | 12% | 88% | 7.33 | Instantaneous |
| 6 | A = N, X = H, Y = H, Z = CHO | 8% | 92% | 11.5 | Instantaneous |
| 7 | A = C, X = CHO, Y = OH, Z = H | 100% | 0% | 0 | 7 days no reaction |

As observed for acetal formation, the regiochemistry of the pyridinium derivatives plays an important role. A slightly larger extent of hydrated product was observed for the compounds in which the aldehyde is at the 2-position compared to the corresponding derivatives with the aldehyde at the 4-position, which suggests that the proximity of the positively charged nitrogen is more important than sterics in this case.
Alkylated pyridinium derivatives
To this point we have explored the relationship between the functional group positioning on the pyridine ring with product formation at neutral and acidic pH. In order to achieve similar reaction rates and extents of addition that we observed under acidic conditions for the formyl pyridine derivatives, we alkylated the pyridine nitrogen (compounds 8 to 11 in Fig. 2). Compound 9 was benzylated due to difficulties encountered in methylating compound 3.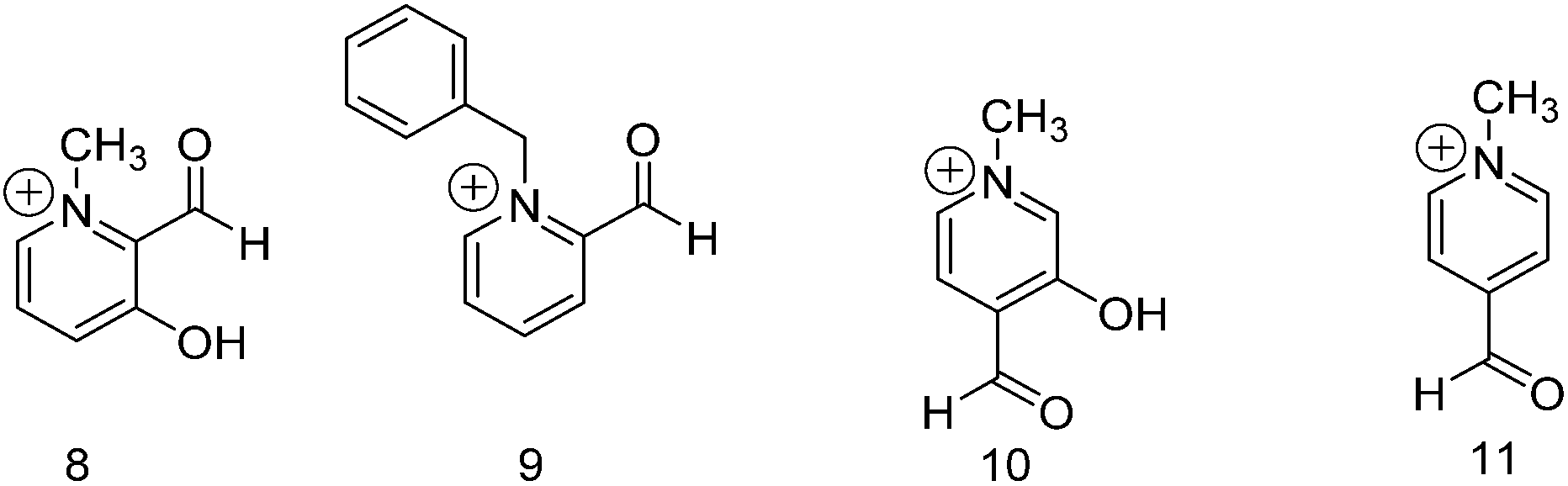 | ||
| Fig. 2 Structures of compounds (8 to 11) used in this study. | ||
Table 5 shows the data for the addition of methanol to compounds 8–11. Compounds 9 and 11 do not form any acetal, but they are converted entirely to hemiacetal within one day. The positively charged pyridine nitrogen activates the aldehyde to attack by the alcohol to form the hemiacetal; however, the oxocarbenium ion intermediate (C in Scheme 2) that would lead to acetal is poorly stabilized without a hydroxyl or methoxy group present. Conversely, in the presence of an adjacent hydroxyl group as in compounds 8 and 10, some acetal is formed due to stabilization of the intermediate by the hydroxyl group. Without including the corresponding methoxy derivatives, it is not possible to conclude whether the effect from the hydroxyl group is primarily due to hydrogen bonding or resonance donation. There is likely a significant population of deprotonated hydroxyl in 8 and 10, which deactivates the aldehyde to hemiacetal formation. Hence, compared to their protonated analogues, compounds 8 and 10 are converted to acetal to a lesser extent probably due to the lower formation of hemiacetal. Nevertheless, more hemiacetal and acetal are formed from 8 and 10 under neutral conditions compared to their pyridine analogues, which demonstrates the potential of this design as a starting point for the development of receptors for alcohols.
|
|||||||
|---|---|---|---|---|---|---|---|
| Compound | Aldehyde | Hemiacetal | Keq (He) | Acetal | Keq (Ac) | Time | |
| 8 | B = Me, X = CHO, Y = OH, Z = H | <1% | 83% | >1000 | 17% | >1000 | 7 days |
| 9 | B = –CH2Ph, X = CHO, Y = H, Z = H | <1% | 100% | >1000 | <1% | Almost 0 | 1 day |
| 10 | B = Me, X = H, Y = OH, Z = CHO | <1% | 38% | >1000 | 62% | >1000 | 7 days |
| 11 | B = Me, X = H, Y = H, Z = CHO | <1% | 100% | >1000 | <1% | Almost 0 | 1 day |
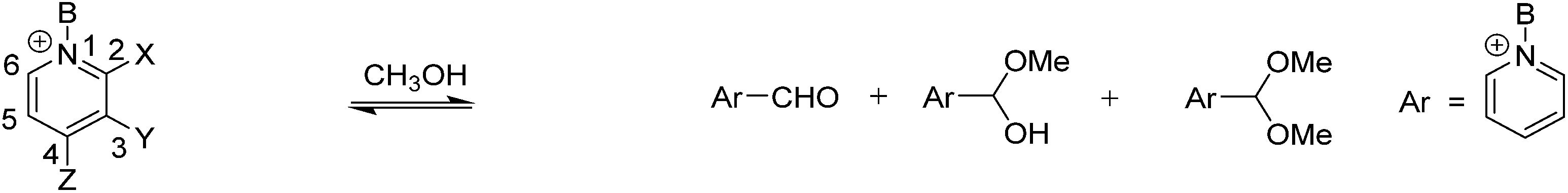
Finally, experiments were carried out to explore the extent of hydrate formation with the four alkylated pyridinium derivatives (8–11, Table 6). Compounds 8–11 behave nearly identically to their protonated counterparts 1, 3, 4, and 6 (see Table 4). This finding shows that alkylating the formyl pyridine compounds is essentially equivalent to protonating them in the case of hydrate formation, and the presence of the hydroxyl anion in 8 and 10 does not appear to have a significant effect.
|
|||||
|---|---|---|---|---|---|
| Compound | Aldehyde | Hydrate | Keq (Hy) | Time | |
| a In the tables, “instantaneous” indicates that the reaction occurred before the 1H-NMR could be taken. | |||||
| 8 | B = Me, X = CHO, Y = OH, Z = H | 14% | 86% | 6.14 | Instantaneous |
| 9 | B = –CH2Ph, X = CHO, Y = H, Z = H | <1% | 100% | >1000 | Instantaneous |
| 10 | B = Me, X = H, Y = OH, Z = CHO | 26% | 74% | 2.85 | Instantaneous |
| 11 | B = Me, X = H, Y = H, Z = CHO | 4% | 96% | 24 | Instantaneous |

Experimental
General
Chemicals were purchased from Sigma Aldrich. The pyridine derivatives were purchased from Matrix Scientific and used as is. The methanol used for the hemiacetal and acetal formation experiments was obtained from a Vacuum Atmospheres Company (VAC) solvent purifier and precautions were taken to keep the methanol moisture free. We ensured the methanol remained dry by keeping it under nitrogen in the presence of 4 Å molecular sieves. Also, the NMR tubes were dried overnight and kept airtight throughout the experimental process; hence, our assumption was that the water generated by the acetal formation was the major contributor to the total water concentration in the final solutions. Since methanol was in large excess and effectively constant, this simplification did not significantly affect our results for the hemiacetal formation reactions. Hence, qualitative data was collected relative to the influence of the hydroxyl group and electron deficient pyridine ring towards the formation of hemiacetal and acetal. The 1H-NMR spectra were recorded on a Varian Unity 300 NMR spectrometer. 1H-NMR spectra of compounds 8, 9, 10, and 11 in methanol and in water are available from Wiley Interscience.Synthesis
The synthesis of N-methylated pyridinium derivatives was carried out by refluxing the pyridine derivatives and methyl iodide in benzene for 24 h (Scheme 3).14–16 The synthesis of an N-benzylated pyridinium derivative was carried out by stirring a solution containing 2-formyl pyridine and benzyl chloride in DMF at 100 °C for 24 h (Scheme 3).17 | ||
| Scheme 3 Synthesis of compounds 8, 10, 11 (top), and 9 (bottom). | ||
Hemiacetal, acetal, and hydrate formation experiments
1H-NMR was used to monitor the reactions. For a quick survey of the reactivity, we monitored the hydrogen in the formyl group and in the subsequent hemiacetal, acetal, and hydrate. An estimate of the time at which a reaction was deemed to be complete was when we took a spectrum and no further signal changes in the characteristic product peaks in the 1H-NMR were observed at room temperature. At that point, we deemed that equilibrium had been reached. The equilibrium constants were calculated with respect to the concentration of the unreacted aldehyde.Conclusions
By studying hemiacetal/acetal formation from formyl pyridine and protonated formyl pyridinium with hydroxyl and methoxy substitution, we were able to probe the role of an adjacent hydroxyl group. For the formyl pyridine derivatives under neutral conditions, the presence of the hydroxyl anion significantly deactivates the carbonyl to alcohol or water addition, while the methoxy group does not appear to significantly deactivate the carbonyl. Also in contrast to the methoxy group, the hydroxyl anion seems to stabilize the oxocarbenium ion intermediate resulting in formation of acetal. For the formyl pyridine derivatives under acidic conditions, the expected trend of resonance donation to stabilize the oxocarbenium ion intermediate is observed: the hydroxyl is a better donor than the methoxy, and the hydrogen has no donor ability. Under acidic conditions, electron donation from the hydroxyl group somewhat deactivates the aldehyde to alcohol or water addition. Furthermore, the steric argument invoked to explain the lower extent of reaction for the ortho derivatives compared to the para derivatives is also not apparent in the cases of the hemiacetal/acetal and hydrate reactions with the pyridinium derivatives. For the pyridinium derivatives, the proximity of the positively charged nitrogen is the dominant effect.Alkylated formyl pyridinium derivatives show promising activity in hemiacetal/acetal formation through alcohol addition to activated carbonyls. The equilibrium for hemiacetal formation was generally reached within a day when no hydroxyl group was present in the alkylated pyridinium derivatives. However, no acetal formation was observed in the absence of a hydroxyl group in the pyridinium ring. This finding demonstrates the importance of the electron donating group in the alkylated pyridinium ring for facilitating acetal formation. It can be concluded that the presence of hydroxyl groups in the alkylated pyridinium derivatives is essential for driving the reaction toward acetal when establishing equilibrium.
Acknowledgements
The authors are grateful for the financial support provided by the National Science Foundation (CHE – 1212971) and the Welch Foundation (F-1151).Notes and references
- E. Breitmaier, Terpenoids and Steroids. Specialist Periodical Reports, ed. K. H. Overton, The Royal Society of Chemistry, London, UK, 1971, vol. 1 CrossRef CAS PubMed
; R. A. Yoder and J. N. Johnston, Chem. Rev., 2005, 105, 4730 CrossRef CAS PubMed
- J. Moreno and R. Peinado, Enological Chemistry, Academic Press, San Diego, 2012 CrossRef CAS PubMed
- R. Nandhakumar, Y. Ahn, H. Yoon and K. Kim, Bull. Korean Chem. Soc., 2009, 30, 2938 CrossRef CAS
- S. L. Wiskur, J. J. Lavigne, H. Ait-Haddou, V. Lynch, Y. H. Chiu, J. W. Canary and E. V. Anslyn, Org. Lett., 2001, 3, 1311 CrossRef CAS PubMed
- H. G. Kuivila, A. H. Keough and E. J. Soboczenski, J. Org. Chem., 1954, 19, 780 CrossRef CAS
- H. Otsuka, E. Uchimura, H. Koshino, T. Okano and K. Kataoka, J. Am. Chem. Soc., 2003, 125, 3493 CrossRef CAS PubMed
- M. Lauer and G. J. Wulff, J. Chem. Soc., Perkin Trans. 2, 1987, 745 RSC
- B. Collins, P. Metola and E. Anslyn, Supramol. Chem., 2012, 1 Search PubMed
- H. Fang, G. Kaur and G. Wang, J. Fluores., 2004, 14, 481 CrossRef CAS
- J. Bell, D. Kubler, P. Sartwell and R. Zepp, J. Org. Chem., 1965, 30, 4284 CrossRef CAS
- M. Matsui, K. Yamada and K. Funabiki, Tetrahedron, 2005, 61, 4671 CrossRef CAS PubMed
- L. You and E. V. Anslyn, Org. Lett., 2009, 11, 5126 CrossRef CAS PubMed
- J. Dragna, G. Pescitelli, L. Tran, V. M. Lynch, E. V. Anslyn and L. DiBari, J. Am. Chem. Soc., 2012, 134, 4398 CrossRef CAS PubMed
- Y. Karube, K. Iwamoto and J. Takata, J. Nucl. Med., 1994, 35, 1691 CAS
- Y. Karube and Y. Matsushima, J. Am. Chem. Soc., 1977, 99, 7356 CrossRef CAS
- W. Sajomsang, P. Gonil and S. Saesoo, Eur. Polym. J., 2009, 45, 2319 CrossRef CAS PubMed
- A. P. Krapcho, ARKIVOC, 2007, 9, 28 Search PubMed
- C. D. Ritchie and W. F. Sager, Prog. Phys. Org. Chem., 1964, 2, 323 CAS
- C. M. Harris, R. J. Johnson and D. E. Metzler, Biochim. Biophys. Acta, 1976, 421, 181 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2014 |