Immobilized oxo-vanadium Schiff base on graphene oxide as an efficient and recyclable catalyst for the epoxidation of fatty acids and esters
Sanny Vermaa,
Mounika Ailab,
Savita Kaul*b and
Suman L. Jain*a
aChemical Sciences Division, CSIR-Indian Institute of Petroleum, Dehradun-248005, India. E-mail: suman@iip.res.in; Fax: +91-135-2660202; Tel: +91-135-2525788
bBiofuel Division, CSIR-Indian Institute of Petroleum, Dehradun-248005, India. E-mail: skaul@iip.res.in
First published on 20th June 2014
Abstract
Oxo-vanadium Schiff base was covalently immobilized onto chemically functionalized graphene oxide (GO) using 3-aminopropyltriethoxysilane as a coupler. The loading of vanadyl Schiff base onto GO nanosheets was confirmed by FTIR, XRD, TGA, and inductively coupled plasma-atomic emission spectroscopy (ICP-AES). The synthesized heterogeneous catalyst was found to be efficient and selective for the epoxidation of fatty acids and esters using t-butyl hydroperoxide (TBHP) as an oxidant. Interestingly, the immobilized catalyst showed a higher catalytic efficiency than the homogeneous vanadyl acetylacetonate. The recycling experiment results indicated that the catalyst was highly stable and maintained very high activity, and selectivity even after being used for six cycles.
Owing to the on-going price increase for fossil resources, their uncertain availability, and due to growing environmental concerns, the biorefinery concept has been developed to integrate the processes and technologies needed to convert biomass into chemicals and fuels.1 Among the known chemicals, epoxidized fatty acids and their derivatives have received considerable interest owing to their widespread application, including as additives in lubricants, as stabilizers in halogen-containing polymers, and in the manufacture of cosmetics, and bioactive chemicals.2 The conventional method for the epoxidation of fatty acid compounds involves the use of peracids which are either pre-formed or synthesized in situ from the reaction of an organic acid, i.e. acetic or formic acid with hydrogen peroxide.3 However, this process for epoxidation has certain drawbacks, such as lower selectivity to the epoxidized products due to the acid-catalyzed ring opening of the epoxide ring, a difficult separation of acidic by-products from the reaction mixture and tedious handling procedures due to the use of highly-concentrated hydrogen peroxide and strong acids. Thus, the development of catalytic processes using inexpensive eco-friendly oxidants is considered to be an attractive approach, from both environmental and economical viewpoints.4 Conventional epoxidation catalysts, including transition metal-based catalysts using hydrogen peroxides and alkyl hydroperoxides, have been extensively used for the epoxidation of unsaturated compounds.5 However, high costs, difficult recovery, and non-recyclability of the catalyst make these catalysts undesirable for large-scale application. As a consequence, many methods and strategies have been reported to immobilize the homogeneous catalysts on insoluble support matrixes such as organic polymers,6 mesoporous silica,7 zeolites8 and clays.9
Recently, graphene oxide (GO), due to the presence of plenty of oxygen functionalities, such as hydroxyl and epoxide groups on both sides of its surface, has been acknowledged as one of the most promising supports to immobilize various homogeneous materials, including transition metal complexes.10 In this regard, Scheuermann et al.11 reported that Pd nanoparticles-supported graphene oxide exhibited higher catalytic activity and low palladium leaching for the Suzuki–Miyaura coupling reaction. Moreover, gold nanoparticles supported on GrO were found to be efficient catalysts for the reduction of o-nitroaniline to 1,2-benzenediamine with significantly higher catalytic activities than the corresponding gold nanoparticles.12 Mungse et al.13 recently reported the grafting of an oxo-vanadium Schiff base onto GrO nanosheets for the oxidation of alcohols, which showed good catalytic behavior.
In a continuation to our work on the development of green oxidation methodologies, herein, we report the synthesis, characterization of an immobilized oxo-vanadium Schiff base onto graphene oxide (VO–NH2-GO), and its use for the epoxidation of fatty acids and esters using t-butyl hydroperoxide as oxidant (Scheme 1). The obtained heterogeneous catalyst VO–NH2-GO exhibited higher catalytic activity than the homogeneous oxo-vanadium catalyst, coupled with the additional benefits of a facile recovery and recycling ability of the catalyst.
 | ||
| Scheme 1 Epoxidation of fatty acids and esters. | ||
GO with a high specific surface area along with plenty of oxygen functionalities decorated on both sides of the nanosheets was used as the support for the immobilization of oxo-vanadium Schiff base using 3-aminopropyltriethoxysilane (APTES) as a coupler. The amino functionalization of the support matrix using APTES for the grafting of metal complexes is well documented in the literature.14 The loading of amino propyl groups onto GO changes the elemental composition with the introduction of nitrogen and silicon. The loading of the amino propyl groups onto the graphene oxide support was found to be 1.5 mmol g−1, as determined by the nitrogen content determined by elemental analysis (2.1% N). Amino-functionalized graphene oxide is labeled as NH2-GO and oxo-vanadium Schiff base immobilized graphene oxide is labeled as VO–NH2-GO. The schematic presentation of the synthesis of heterogeneous vanadyl complex is shown in Scheme 2.
 | ||
| Scheme 2 Schematic presentation of the synthesis of oxo-vanadium Schiff base immobilized to graphene oxide (VO–NH2-GO). | ||
The BET surface area of synthesized GO, NH2-GO and VO–NH2-GO was found to be 85, 27, and 22.8 m2 g−1, respectively.
The chemical changes that occurred during the immobilization of vanadyl acetylacetonate complex onto GO were examined by FTIR (Fig. 1), showing the vibrational assessments of GO (Fig. 1a), NH2-GO (Fig. 1b) and VO–NH2-GO (Fig. 1c). The strong bands at 1731, 1222.9, and 1035.5 cm−1, were attributed to the stretching modes of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–OH, and C–O–C, respectively, revealing the presence of carboxyl, and epoxy groups in pure GO.15,16 In addition, a broad peak at 3392 cm−1 is attributed to the stretching mode of O–H bonds, revealing the presence of many hydroxyl groups. The peak at 1629 cm−1 is attributed to the CC stretching of sp2-bonded carbons in the graphene oxide.13 The FTIR spectrum of NH2-GO exhibited a doublet at 2850 cm−1 and 2917 cm−1, corresponding to the symmetric CH2 and asymmetric CH2 of the alkyl chains assigned to the silane moieties of APETS, and confirms the successful grafting of aminosilane moieties onto the GO support. Moreover, the appearance of bands at 1108 cm−1, 1037 cm−1 and 1620 cm−1 due to Si–O–Si, Si–O–C, and C–N vibration, respectively,17 provided more evidence for the successful chemical functionalization.18 The FTIR spectra of VO–NH2-GO shows a new peak at around 1610 cm−1, attributed to CN stretching due to the Schiff base, indicating that the chemical surroundings of the nitrogen atom have been changed and almost all of the –NH2 groups have reacted with acetylacetone to form a Schiff base.19 Moreover, peaks in the region of 1600–1300 cm−1 are due to C–O and C–N vibrations, indicating that the Schiff base was successfully immobilized onto GO.
O, C–OH, and C–O–C, respectively, revealing the presence of carboxyl, and epoxy groups in pure GO.15,16 In addition, a broad peak at 3392 cm−1 is attributed to the stretching mode of O–H bonds, revealing the presence of many hydroxyl groups. The peak at 1629 cm−1 is attributed to the CC stretching of sp2-bonded carbons in the graphene oxide.13 The FTIR spectrum of NH2-GO exhibited a doublet at 2850 cm−1 and 2917 cm−1, corresponding to the symmetric CH2 and asymmetric CH2 of the alkyl chains assigned to the silane moieties of APETS, and confirms the successful grafting of aminosilane moieties onto the GO support. Moreover, the appearance of bands at 1108 cm−1, 1037 cm−1 and 1620 cm−1 due to Si–O–Si, Si–O–C, and C–N vibration, respectively,17 provided more evidence for the successful chemical functionalization.18 The FTIR spectra of VO–NH2-GO shows a new peak at around 1610 cm−1, attributed to CN stretching due to the Schiff base, indicating that the chemical surroundings of the nitrogen atom have been changed and almost all of the –NH2 groups have reacted with acetylacetone to form a Schiff base.19 Moreover, peaks in the region of 1600–1300 cm−1 are due to C–O and C–N vibrations, indicating that the Schiff base was successfully immobilized onto GO.
 | ||
| Fig. 1 FTIR spectra of: (a) GO; (b) NH2-GO; (c) VO–NH2-GO. | ||
XRD studies
XRD patterns are used to study the changes in structure of graphene oxide after the immobilization of the oxo-vanadium complex (Fig. 2). The XRD spectrum of GO, as shown in Fig. 2a, shows the characteristic diffraction peak at 10.8°, corresponding to (001), which is due to the oxidation of graphite powder leading to the introduction of various oxygen containing functional groups.20 After amino functionalization and immobilization of the oxo-vanadium Schiff base, this peak disappeared and another broad diffraction of graphite (002) at 2θ value ∼22° appeared, which is closer to the reduced graphene, indicating that the major oxygen containing groups of GO have been successfully functionalized.21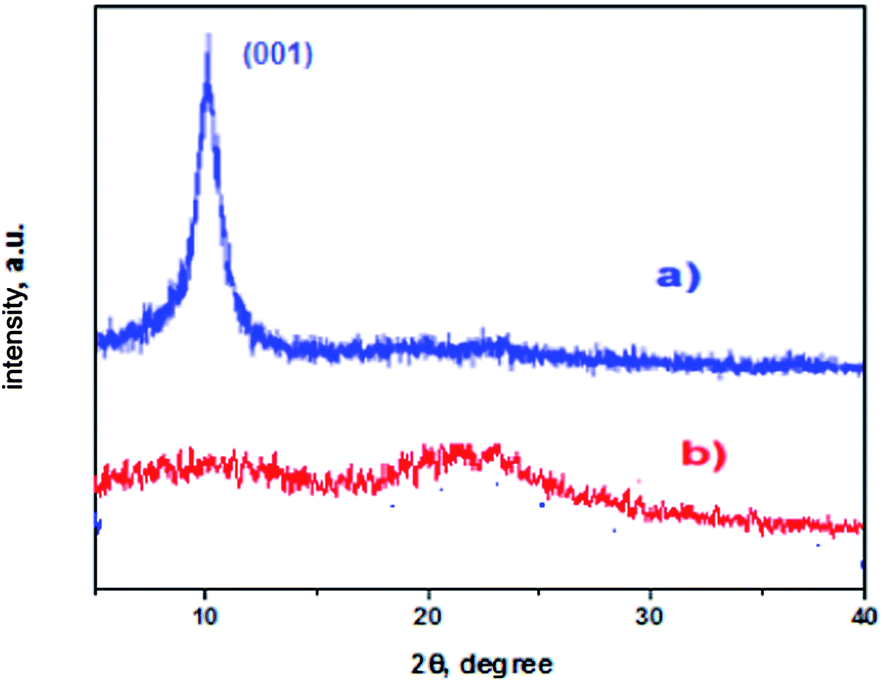 | ||
| Fig. 2 XRD pattern of (a) GO; (b) VO–NH2-GO. | ||
SEM studies
The SEM images of GO and VO–NH2-GO are shown in Fig. 3a and b. It can be seen that GO and VO–NH2-GO have a similar appearance, having two-dimensional nanosheets with wave-like structures.22 Furthermore, EDAX analysis (Fig. 3c) confirmed the presence of vanadium in the synthesized heterogeneous catalyst. | ||
| Fig. 3 SEM image of (a) GO; (b) VO–NH2-GO; (c) EDAX of VO–NH2-GO. | ||
The TG curves of GO and VO–NH2-GO are depicted in Fig. 4. The samples were heated at a constant rate of 10 °C min−1 from 100 °C to 800 °C under N2 flow. The TG curve of GO shows the first weight loss in the range of 100–150 °C, evidently owing to the evaporation of water molecules trapped in the material. The second and significant weight loss was observed in the range of 150–250 °C and is assigned to the thermal decomposition of labile oxygen-containing functional groups. The VO–NH2-GO catalyst exhibited three mass losses up to 550 °C.23 The first weight loss occurred between 100 and 250 °C evidently owing to the evaporation of physically adsorbed water. The second weight loss in the temperature range from 250 to 450 °C is attributed to the thermal decomposition of undigested oxygen carrying functionalities and APTMS moieties, which have not participated in further chemical functionalization. The last major mass loss observed in the range of 360–470 °C is related to the slow decomposition of the VO-Schiff base complex.24 Based on the weight loss during the thermal decomposition of VO–NH2-GO, the loading of Schiff base ligand in the synthesized catalyst was found to be 0.52 mmol g−1. However, the vanadium content as estimated by ICP-AES was found to be 0.24 mmol g−1, indicating that the molar ratio of the Schiff base and vanadium in the synthesized catalyst is nearly 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Based on the nitrogen content (0.76% N) in the synthesized catalyst as determined by elemental analysis, the loading of oxo-vanadium Schiff base was found to be 0.54 mmol g−1. This value is in good agreement with the TGA results.
:1. Based on the nitrogen content (0.76% N) in the synthesized catalyst as determined by elemental analysis, the loading of oxo-vanadium Schiff base was found to be 0.54 mmol g−1. This value is in good agreement with the TGA results.
 | ||
| Fig. 4 TGA curve of (a) GO; (b) VO–NH2-GO. | ||
Catalytic activity
The synthesized material was used as a catalyst for the oxidation of fatty acids and their esters to the corresponding epoxides using t-butyl hydroperoxide as the oxidant without any additional organic solvent. The experiments were carried out batchwise in a 25 ml roundbottomed flask equipped with a magnetic stirrer and reflux condenser. In order to obtain the optimum reaction condition, various parameters (reaction time, molar ratio of substrate to TBHP, temperature and catalyst concentration) influencing its catalytic property were studied using oleic acid as the representative substrate. The results of these optimization experiments are summarized in Table 1.| Entry | Time (h) | Molar ratio of oleic acid: TBHP | Temp. (°C) | Catalyst conc. (mg) | Conversion (%) | Selectivity for epoxide (%) |
|---|---|---|---|---|---|---|
| 1 | 2 | 1:1.5 |
65 | 30 | 48.8 | 99.0 |
| 2 | 4 | 1:1.5 |
65 | 30 | 80.5 | 99.1 |
| 3 | 5 | 1:1.5 |
65 | 30 | 98.0 | 99.1 |
| 4 | 5 | 1:1 |
65 | 30 | 58.8 | 98.7 |
| 5 | 5 | 1:2.0 |
65 | 30 | 98.4 | 99.2 |
| 6 | 5 | 1:2.5 |
65 | 30 | 98.5 | 97.4 |
| 7 | 5 | 1:1.5 |
25 | 30 | 42.5 | 99.1 |
| 8 | 5 | 1:1.5 |
40 | 30 | 60.4 | 99.0 |
| 9 | 5 | 1:1.5 |
80 | 30 | 99.0 | 55.7 |
| 10 | 5 | 1:1.5 |
65 | 20 | 88.4 | 99.0 |
| 11 | 5 | 1:1.5 |
65 | 50 | 99.8 | 99.1 |
The reaction time is found to show a significant effect on the conversion of the oleic acid to the corresponding epoxide. It is observed that the conversion of oleic acid gradually increases from 2 to 5 h. However, the reaction time has little effect on the epoxide selectivity (Table 1, entries 1–3).
The effect of the molar ratio of the ethylene unsaturation/TBHP ratio on the conversion to epoxide was studied by varying the ratio from 1:1 to 1:2.5 (Table 1, entries 4–6). In all cases, we obtained very high conversion and high selectivity for the epoxide formation. This reflects that the TBHP concentration does not affect the reaction much and that the developed method is more highly selective for the epoxide formation than for CC bond cleavage.
Reaction temperature: to determine the optimum reaction temperature for the epoxdiation reaction, a series of experiments was performed by employing the described procedure and varying the reaction temperature (Table 1, entries 7–9). The temperature has a remarkable effect on the selectivity of epoxide. Amongst the three tested temperatures of 25, 40 and 65 °C, a maximum conversion of oleic acid and a maximum selectivity to epoxide was obtained at 65 °C. A further increase in the reaction temperature (to 80 °C) reduced the epoxide selectivity significantly. Therefore, we considered 65 °C as the optimum temperature for the present study.
Catalyst amount: to find the optimum catalyst concentration, a series of epoxidation reactions of oleic acid was performed in the presence of graphene oxide immobilized oxo-vanadium catalyst, by varying its amount in the range of 20–50 mg at 65 °C (Table 1, entries 10–11). The epoxide yield was found to increase as the catalyst concentration increased from 20 to 30 mg. A further increase in the catalyst concentration does not influence the epoxide yield significantly. This is probably due to the reason that every reaction achieves its optimum catalyst concentration value. Beyond that value, excessive catalyst does not affect the reaction rate to any significant extent.25 Hence, the epoxidation reactions were carried out by employing 30 mg of catalyst as the optimum amount.
Based on these results, the optimized reaction conditions for the epoxidation of fatty acid and esters should be: catalyst (30 mg), substrate/TBHP (1:1.5), time 5 h and temperature 65 °C. The reaction was further generalized to a variety of mono- and poly-unsaturated fatty acids and esters. The results of these experiments are summarized in Table 2. All the substrates were efficiently converted to the corresponding epoxides with excellent selectivity under the described experimental conditions. The formation of the epoxide product was initially characterized by the FTIR technique. The disappearance of the band at 3005 cm−1 because of the –CC– of the double bond in the FTIR spectra of epoxidized products supports the consumption of double bonds, and the formation of epoxides during the reaction. Furthermore, in the 1H NMR spectra, the peak between 5.3–5.5 ppm due to the protons attached to –CC– disappeared, confirming the successful formation of epoxides. In addition, the appearance of a multiplet in the range of 3.1–3.2 ppm because of epoxide protons confirmed the formation of the respective epoxidized products. Unreacted peroxide was determined by iodometric titration. Blank experiments, in the absence of catalyst, did not produce the epoxidized product even after a prolonged reaction time (12 h). Similarly, the oxidation of oleic acid was performed using homogeneous vanadyl acetylacetonate under the described experimental conditions. The reaction using homogeneous catalyst was found to be slow, affording a lesser conversion of oleic acid (92.5% with 96.5% selectivity of the epoxide) than heterogeneous vanadium catalyst. The superior catalytic activity of the heterogeneous catalyst can be attributed to the promoting effect of the graphene oxide support containing plenty of oxygen functionalities, which may be facilitating the reaction via interactions with the substrate and TBHP on the surface.
| Entry | Substrate | Product | Conv.b (%) | EO selec.b (%) | Yieldc (%) |
|---|---|---|---|---|---|
| a Reaction conditions: substrate (1 mmol), molar ratio of substrate and TBHP (1:1.5), catalyst (30 mg), temperature 65 °C, reaction time 5 h.b The conversion and selectivity for the epoxide were determined by GC-MS.c Isolated yield; EO = epoxide. |
|||||
| 1 |  |
 |
98.0 | 99.1 | 97 |
| 2 |  |
 |
95.4 | 99.0 | 94 |
| 3 |  |
 |
99 | 99 | 97 |
| 4 |  |
 |
80 | 98.4 | 78 |
| 5 |  |
 |
98 | 99.1 | 96 |
| 6 |  |
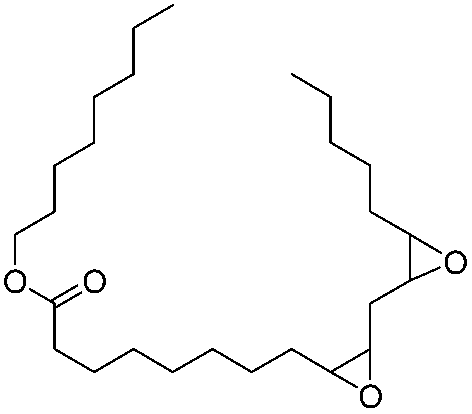 |
99 | 99.2 | 97 |
| 7 |  |
 |
98 | 99.0 | 96 |
| 8 |  |
 |
98 | 99.1 | 97 |
To verify the scalability of the process, the oxidation of oleic acid was performed in a higher mmol (20 mmol) under identical experimental conditions (see Experimental section). The conversion of oleic acid was found to be 98% with 99% selectivity for the epoxide. However, the isolated yield of the product was found to be 97.4%. These findings suggested that the developed method can be used to scale up the process.
Reusability of the heterogeneous catalyst: reusability of the solid catalyst is one of the most important criteria to make it viable at an industrial scale. To study the reusability of graphene oxide immobilized vanadium catalyst, the epoxidation of oleic acid was performed by following the described procedure. After the reaction, the catalyst was recovered from the reaction mixture by filtration, washed with ethanol, and then dried for 24 h. The recovered catalyst was used for six consecutive runs by following the same experimental conditions. The reused catalyst was found to be equally active and afforded almost a similar conversion and selectivity of the epoxide, as shown in Table 3. These results confirmed that the developed catalyst is highly stable and can be recycled without significant loss in catalytic activity. Importantly, no leaching of the metal could be detected during the experiments, as ascertained by ICP-AES analyses, indicating that the reactions were truly heterogeneous in nature.
Oxo-vanadium Schiff base complex was immobilized onto graphene oxide support previously functionalized with 3-aminopropyltriethoxysilane. The prepared catalyst was characterized by FTIR, XRD, SEM, TGA and ICP-AES. The immobilized oxo-vanadium catalyst was found to be an efficient and highly selective catalyst for the epoxidation of fatty acids and esters using TBHP as an oxidant. In addition, developed heterogeneous catalyst was found to be quite stable during the reaction and could be reused for six subsequent runs. The leaching studies showed that there was no loss of vanadium during the reaction, and the vanadium content in fresh and recovered catalyst was found to be almost similar, as ascertained by ICP analysis. These results suggested that the graphene oxide immobilized oxo-vanadium Schiff base catalyst was truly heterogeneous in nature.
Techniques used
Fourier Transform Infrared Spectra were recorded on a Perkin-Elmer spectrum RX-1 FTIR spectrophotometer using a potassium bromide window. Scanning electron microscopy (SEM) images of the synthesized GO and modified GO were executed using FE-SEM (Jeol Model JSM-6340F). X-ray powder diffraction patterns were recorded on a Bruker D8 Advance diffractometer at 40 kV and 40 mA with Cu Kα radiation (λ = 0.15418 nm) for confirming GrO synthesis and its functionalization, and samples for XRD were prepared on a glass slide by adding well dispersed catalyst into the slot and drying properly. The surface properties like Brunauer–Emmet–Teller (BET) surface area, Barret–Joyner–Halenda (BJH) porosity, pore volume of GO and modified GO were examined by N2 adsorption–desorption isotherms at 77 K by using VP; Micromeritics ASAP2010. Thermo gravimetric analyses (TGA) of the samples were carried out using a thermal analyzer TA-SDT Q-600. Analysis was carried out in the temperature range of 40–800 °C under a nitrogen flow at a heating rate 10 °C min−1. For determining vanadium loading in the synthesized catalyst, ICP-AES analyses were carried out by an inductively coupled plasma-atomic emission spectrometer (ICP-AES, DRE, PS-3000UV, Leeman Labs Inc, USA). Metal in 0.05 g catalyst was leached out using conc. HNO3, and the final volume was made up to 10 ml by adding distilled water.Synthesis of graphene oxide
Graphene oxide (GO) was synthesized from graphite flakes using the modified Hummers method.26 In a typical experiment, 46 ml concentrated H2SO4 was added into a flask containing graphite flakes (2 g) and sodium nitrate (0.75 gm) and the resulting mixture was cooled down to 0 °C in an ice bath under stirring. Then, KMnO4 (6.0 g) was added slowly to this mixture at 0 °C and the mixture was stirred for 5 days. 100 ml diluted H2SO4 (5 wt%) was added to this and heated at 90 °C for 2 h with continuous stirring. A subsequent addition of 30 wt% H2O2 solution (approximately 5.4 ml), followed by stirring for 2 h at room temperature, afforded graphene oxide, which was collected by centrifugation. The as-synthesized material was subsequently washed with by 5% HCl and deionized water and dried at 70 °C under vacuum.Synthesis of amino-functionalized graphene oxide (NH2-GO)
The synthesized graphene GO (1.0 g, 30 ml) was taken in anhydrous toluene, and then 3-aminopropyltrimethoxysilane (3-APTES, 2.5 mmol) was added under a N2 atmosphere. The resultant suspension was refluxed at 110 °C under a nitrogen atmosphere for 24 h. The resultant solid was washed with toluene three times, and then the resultant amino-functionalized grapheme oxide (NH2-GO) was dried under vacuum oven at 50 °C for 24 h.Immobilization of oxo-vanadium Schiff base to amino functionalized graphene oxide (VO–NH2-GO)
NH2-GO (1 g) was dispersed in ethanol (30 ml), and then VO(acac)2 (200 mg) was added. The mixture was heated at 65 °C for 3.5 h. After cooling the reaction mixture to room temperature, the heterogeneous graphene oxide immobilized catalyst was separated by filtration and subsequently washed with ethanol to remove the unreacted VO(acac)2. The resultant solid powder was dried at 50 °C under vacuum and labeled as VO–NH2-GO. The loading of vanadium in the synthesized catalyst was found to be 0.23 mmol g−1, as determined by ICP-AES analysis.Typical experimental procedure for the oxidation of fatty acids and esters
The epoxidation of fatty acids and esters was carried out in a 25 ml round-bottomed flask equipped with a magnetic stirrer and reflux condenser. In a typical experiment, oleic acid (1 mmol), anhydrous TBHP in dodecane (1.5 mmol) and catalyst (30 mg) were added to the flask, and the resultant mixture was continuously stirred at 65 °C. After reaction, the solid catalyst was separated by filtration, washed with ethanol, and then dried at 50 °C for 24 h. The recovered catalyst was used in order to investigate the stability and recyclability. The products of the epoxidation were analyzed by a gas using a Varian capillary column CP SIL 24CB and a FID detector. The conversion and selectivity of the product was determined by GC and the identity of the product was established by comparing its spectral data with the authentic sample.Experimental procedure for scale up the process
Oleic acid (20 mmol), anhydrous TBHP in dodecane (30 mmol) and catalyst (600 mg) were added to the 250 ml round-bottomed flask, and the resultant mixture was continuously stirred at 65 °C for 5 h. After completion of the reaction, the solid catalyst was separated by filtration, washed with ethanol, and dried at 50 °C for 24 h. The conversion and selectivity of the product was determined by GC and the identity of the product was established by comparing its spectral data with the authentic sample. The conversion of oleic acid was found to be 98% with 99% selectivity for the epoxide. However, the isolated yield of the product was found to be 97.4%.Acknowledgements
We kindly acknowledge Director IIP for his kind permission to publish these results. Analytical Science Division of the Institute is acknowledged for providing analyses of the samples. SV is thankful to CSIR, New Delhi for his research fellowship. MA is also thankful to CSIR, New Delhi for working as Technical HR in XII Five Year project.Notes and references
- C. Briens, J. Piskorz and F. Berruti, Int. J. Chem. React. Eng., 2008, 6, R2, DOI:10.2202/1542-6580.1674.
- (a) U. Biermann, W. Friedt, S. Lang, W. Luhs, G. Machmuller, J. O. Metzger, M. R. Klaas, H. J. Schafer and M. P. Schneider, Angew. Chem., Int. Ed., 2000, 39, 2206–2224 CrossRef CAS; (b) S. Tayde, M. Patnaik, S. L. Bhagt and V. C. Renge, Int. J. Adv. Eng. Tech., 2011, II, 491–501 Search PubMed.
- (a) S. Dinda, A. V. Patwardhan, V. V. Goud and N. C. Pradhan, Bioresour. Technol., 2008, 99, 3737–3744 CrossRef CAS PubMed; (b) C. Cai, H. Dai, R. Chen, C. Su, X. Xu, S. Zhang and L. Yang, Eur. J. Lipid Sci. Technol., 2008, 110, 341–346 CrossRef CAS; (c) P. Lathi and B. Mattiasson, Appl. Catal., B, 2007, 69, 207–212 CrossRef CAS PubMed.
- (a) M. Guidotti, N. Ravasio, R. Psaro, E. Gianotti, L. Marchese and S. Coluccia, Green Chem., 2003, 5, 421–424 RSC; (b) A. Campanella, C. Fontanini and M. A. Baltans, Chem. Eng. J., 2008, 144, 486–492 CrossRef PubMed; (c) Y. Ishii, K. Yamawaki, T. Ura, H. Yamada, T. Yoshida and M. Ogawa, J. Org. Chem., 1988, 53, 3587–3593 CrossRef CAS; (d) W. A. Herrman, J. D. G. Correia, F. E. Khün, G. R. J. Artus and C. C. Rao, Chem.–Eur. J., 1996, 2, 168–173 CrossRef; (e) M. Guidotti, N. Ravasio, R. Psaro, E. Gianotti, L. Marchese and S. Coluccia, Green Chem., 2003, 5, 421–424 RSC.
- (a) X.-T. Zhou, Q.-H. Tang and H.-B. Ji, Tetrahedron Lett., 2009, 50, 6601–6605 CrossRef CAS PubMed; (b) G. Grivani, G. Bruno, H. A. Rudbari, A. D. Khalaji and P. Pourteimouri, Inorg. Chem. Commun., 2012, 18, 15–20 CrossRef CAS PubMed; (c) M. G. Topuzova, S. V. Kotov and T. M. Kolev, Appl. Catal., A, 2005, 281, 157–166 CrossRef CAS PubMed.
- (a) G. Grivani, S. Tangestaninejad, M. H. Habibi, V. Mirkhani and M. Moghadam, Appl. Catal., A, 2006, 299, 131–136 CrossRef CAS PubMed; (b) C. E. Song, E. J. Roh, B. M. Yu, D. Y. Chi, S. C. Kim and K. J. Lee, Chem. Commun., 2000, 615–616 RSC; (c) L. Canali, E. Cowan, H. D. Deleuze, C. L. Gibson and D. C. Sherrington, J. Chem. Soc., Perkin Trans. 1, 2000, 2055 RSC.
- (a) Y. Yang, H. Ding, S. Hao, Y. Zhang and Q. Kan, Appl. Organomet. Chem., 2011, 25, 262–269 CrossRef CAS; (b) G.-J. Kim and J.-H. Shin, Tetrahedron Lett., 1999, 40, 6827–6830 CrossRef CAS.
- (a) Y. Yang, H. Ding, S. J. Hao, Y. Zhang and Q. B. Kan, Appl. Organomet. Chem., 2011, 25, 262–269 CrossRef CAS; (b) I. L. Viana Rosa, C. M. C. P. Manso, O. A. Serra and Y. Iamamoto, J. Mol. Catal. A: Chem., 2000, 160, 199–205 Search PubMed.
- R. I. Kureshy, N. H. Khan, S. H. R. Abdi, I. Ahmad, S. Singh and R. V. Jasra, J. Catal., 2004, 221, 234–240 CrossRef CAS PubMed.
- (a) T. Szabo, O. Berkesi, P. Forgo, K. Josepovits, Y. Sanakis, D. Petridis and I. Dekany, Chem. Rev., 2006, 18, 2740–2749 CAS; (b) Y. Hu, H. Zhang, P. Wu, H. Zhang, B. Zhou and C. Cai, Phys. Chem. Chem. Phys., 2011, 13, 4083–4094 RSC; (c) C. V. Rao, C. R. Cabrera and Y. Ishikawa, J. Phys. Chem. C, 2011, 115, 21963–21970 CrossRef; (d) S. Guo, S. Dong and E. Wang, ACS Nano, 2010, 4, 547–555 CrossRef CAS PubMed; (e) S. Guo and S. Sun, J. Am. Chem. Soc., 2012, 134, 2492–2495 CrossRef CAS PubMed.
- (a) G. M. Scheuermann, L. Rumi, P. Steurer, W. Bannwarth and R. Mulhaupt, J. Am. Chem. Soc., 2009, 131, 8262–8270 CrossRef CAS PubMed; (b) J. Huang, L. Zhang, B. Chen, N. Ji, F. Chen, Y. Zhang and Z. Zhang, Nanoscale, 2010, 2, 2733–2738 RSC.
- C. Yuan, W. Chen and L. Yan, J. Mater. Chem., 2012, 22, 7456–7460 RSC.
- H. P. Mungse, S. Verma, N. Kumar, B. Sain and O. P. Khatri, J. Mater. Chem., 2012, 22, 5427–5433 RSC.
- (a) N. Anand, K. H. P. Reddy, K. S. R. Rao and D. R. Burri, Catal. Lett., 2011, 141, 1355–1363 CrossRef CAS PubMed; (b) N. Anand, K. H. P. Reddy, V. Swapna, K. S. R. Rao and D. R. Burri, Microporous Mesoporous Mater., 2011, 143, 132–140 CrossRef CAS PubMed; (c) C. K. P. Neeli, S. Ganji, V. S. P. Ganjala, S. R. R. Kamarajua and D. R. Burri, RSC Adv., 2014, 4, 14128–14135 RSC.
- A. Stuart, Infrared Spectroscopy: Fundamentals and Applications, J. Wiley, Chichester, West Sussex, England, Hoboken, NJ, 2004 Search PubMed.
- (a) J. I. Paredes, S. Villar-Rodil, A. Martinez-Alonso and J. M. D. Tascon, Langmuir, 2008, 24, 10560–10564 CrossRef CAS PubMed; (b) R. Pena-Alonso, F. Rubio, J. Rubio and J. L. Oteo, J. Mater. Sci., 2007, 42, 595–603 CrossRef CAS PubMed; (c) R. M. Pasternack, S. R. Amy and Y. J. Chabal, Langmuir, 2008, 24, 12963–12971 CrossRef CAS PubMed.
- Y. Gao, Y. Zhang, C. Qiu and J. Zhao, Appl. Organomet. Chem., 2011, 25, 54–60 CrossRef CAS.
- M. Santos, F. E. Kuhn, W. M. Xue and E. Herdtweck, J. Chem. Soc., Dalton Trans., 2000, 3570–3574 RSC.
- S. Dorbes, C. Pereira, M. Andrade, D. Barros, A. M. Pereira, S. L. H. Rebelo, J. P. Araujo, J. Pires, A. P. Carvalho and C. Freire, Microporous Mesoporous Mater., 2012, 160, 67–74 CrossRef CAS PubMed.
- Y. H. Zu, J. Y. Tang, W. C. Zhu, M. Zhang, G. Liu, Y. Liu, W. X. Zhang and M. J. Jia, Bioresour. Technol., 2011, 102, 8939–8944 CrossRef CAS PubMed.
- D. Pan, S. Wang, B. Zhao, M. Wu, H. Zhang, Y. Wang and Z. Jiao, Chem. Mater., 2009, 21, 3136–3142 CrossRef CAS.
- (a) S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS PubMed.
- W. A. Herrmann, W. M. Wachter, F. E. Kühn and R. W. Fischer, J. Organomet. Chem., 1998, 553, 4432–4436 CrossRef.
- H. Su, Z. Li, Q. Huo, J. Guan and Q. Kan, RSC Adv., 2014, 4, 9990–9996 RSC.
- (a) P. Visuvamithiran, M. Palanichamy, K. Shanthi and V. Murugesan, Appl. Catal., A, 2013, 462, 31–38 CrossRef PubMed; (b) S. B. Kumar, S. P. Mirajkar, G. C. G. Pais, P. Kumar and R. Kumar, J. Catal., 1995, 156, 163–166 CrossRef CAS; (c) H. Yang, G. Wei, X. Wang, F. Lin, J. Wang and M. Shen, Catal. Commun., 2013, 39, 5–9 CrossRef PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |