
Surfactant-free platinum nanocubes with greatly enhanced activity towards methanol/ethanol electrooxidation†
Abstract
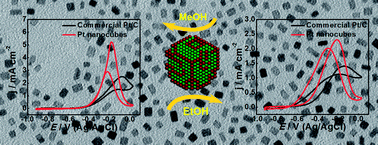
This communication demonstrates a facile approach to synthesize a platinum (Pt) nanocube catalyst (∼3.5 nm) in the absence of surfactants, which exhibits excellent activity towards alcohol electrooxidation. The clean and unique surface structure was proposed as the main reason accounting for the enhancement of catalytic activities.
 Please wait while we load your content...
Please wait while we load your content...

