
Synthesis, spectroscopic characterization, pH dependent redox mechanism and DNA binding behavior of chlorohydroxyaniline derivatives†
Abstract
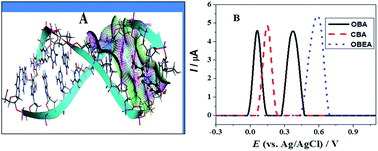
The derivatives of anilines are promisingly useful in rechargeable batteries, electrochromics and biosensors. Phenol and aniline based compounds are bestowed with strong antioxidant and anticancer activities. Based on these considerations three new chlorohydroxyanilines (CHAs) were synthesized and characterized by IR, 13C NMR, 1H NMR and UV-Vis spectroscopy. Cyclic, differential pulse and square wave voltammetry were used for the investigations of the electrochemical fate of these compounds in different pH media. Computational chemistry was used as a tool to verify the experimental outcomes. Two chlorohydroxyanilines were found to oxidize at a potential lower than the standard antioxidant, ascorbic acid. The pH dependent oxidation indicated the involvement of protons during electron transfer reactions. The quasi-reversible and irreversible nature of the first and second oxidation peak was evidenced by square wave voltammetry. The slope of peak potential vs. pH plot and the width at half peak height indicated 1e and 1H+ involvement in each oxidation step. Molecular docking and UV-Vis spectroscopy revealed that all chlorohydroxyanilines interact with DNA via an electrostatic binding mode. A sensitive differential pulse technique allowed the determination of a very low limit of detection.
 Please wait while we load your content...
Please wait while we load your content...

