Minimization of electrical energy consumption in the photocatalytic reduction of Cr(VI) by using immobilized Mg, Ag co-impregnated TiO2 nanoparticles†
Hamed Eskandarlooa,
Alireza Badiei*a,
Mohammad A. Behnajadyb and
Ghodsi Mohammadi Ziaranic
aSchool of Chemistry, College of Science, University of Tehran, Tehran, Iran. E-mail: abadiei@ut.ac.ir; Fax: +98-2161113301; Tel: +98-2161112614
bDepartment of Chemistry, College of Science, Tabriz Branch, Islamic Azad University, Tabriz, Iran
cDepartment of Chemistry, Faculty of Science, Alzahra University, Tehran, Iran
First published on 18th June 2014
Abstract
Magnesium and silver co-impregnated TiO2 nanoparticles were immobilized on a glass plate and used as a fixed-bed system for photocatalytic reduction of Cr(VI) to the less harmful Cr(III). Response surface methodology as a statistical technique was employed for optimizing the preparation conditions of Mg–Ag/TiO2, focusing on minimization of electrical energy consumption as the most important factor in selecting a wastewater treatment technology. Results showed that maximum photocatalytic reduction (84.44%), with minimum electrical energy consumption (30.31 kW h per m3 per order) were achieved at the optimized preparation conditions: Mg content of 0.82 wt%, Ag content of 2.6 wt%, and calcination temperature of 495 °C, whereas at the same conditions, using pure TiO2, Mg/TiO2, and Ag/TiO2 samples lead to 21.17%, 32.59%, and 63.61% photocatalytic reduction rates, and 232.1, 152.03, and 71.42 kW h per m3 per order electrical energy consumptions, respectively. The optimized Mg, Ag co-impregnated TiO2 nanoparticles were characterized by using XRD, SEM, TEM, DRS, EDX, and PL techniques. The considerable PL quenching in the co-impregnated TiO2 with optimized metals content suggests that the co-impregnation of Mg and Ag onto TiO2 could effectively inhibit the recombination probability of photogenerated electrons–holes pairs. Co-impregnation of Mg and Ag metals, and optimization of preparation conditions provides a synergistic effect in enhancement of the TiO2 activity and effective minimization of the electrical energy consumption and treatment cost.
1. Introduction
Recently, removal of heavy metals like chromium has received considerable attention because of their health problems. Industries typically use chromium in various processes like chromate preparation, electroplating, textile industries, leather tanning, paint, and metal finishing.1,2 Chromium exists in two oxidation states as trivalent (Cr(III)) and hexavalent (Cr(VI)) forms. Cr(VI) is more soluble and more toxic (500 times) than the Cr(III) form. Cr(VI) has been reported to be toxic to animals and humans, and is known to be carcinogenic with high mobility in the environment, and the World Health Organization (WHO) recommends that the levels of Cr(VI) in water should be reduced to 0.005 mg L−1.3–5 Many techniques have been used for the removal of Cr(VI) from aqueous media such as adsorption,6,7 electrochemical precipitation,8,9 reverse osmosis,10 solvent extraction,11 ion exchange,12,13 and foam flotation.14,15 However, most of these techniques required either large quantities of chemicals and high energy, and in some of these techniques Cr(VI) is only transferred from one phase to another,16,17 whereas the heterogeneous photocatalytic process is found to be an efficient and clean technology for Cr(VI) reduction.18–20Heterogeneous photocatalysis processes, based on a combination of UV irradiation and a metal oxide (such as TiO2, ZnO, etc.), have received considerable attention in the removal of toxic metals from aqueous media.21 Titania is the most commonly employed metal oxide in the photocatalytic treatment of pollutants, due to its efficient photocatalytic properties, chemical stability, accessibility, low cost, and non-toxicity.22 When TiO2 absorbs a photon with energy greater than or equal to the band gap energy, valence band electrons are promoted to the conduction band leaving holes behind.23 These electron–hole pairs on the surface of TiO2, can reduce or oxidize species in solution having suitable redox potentials.24,25 In the case of Cr(VI) ion removal with this method, the Cr(VI) ion is reduced on the TiO2 surface and converted to a less harmful Cr(III), which can then be precipitated in neutral or alkaline solutions.26,27
The quick recombination of photoinduced charge carriers decreases the photocatalytic efficiency of TiO2.28 One possible solving route to prevent the recombination of charge carriers, is impregnation of TiO2 with metal elements. The metal nanoparticles on the surface of TiO2 will act as an electron reservoir to trap electrons which can greatly increase the efficiency of charge separation, resulting in the improvement of the photocatalytic activity.29–31 However, recently has been reported that the impregnation of TiO2 with a metal element has not been found to meet practical applications and co-impregnation with different metal elements may lead to synergistic effect in the enhancement of photocatalytic activity.21,32–34 A literature review revealed that the impregnation of TiO2 nanoparticles with silver and magnesium metals have attracted significant attention because of considerable photocatalytic activity of this catalysts.35–40
A major fraction of the operating costs in heterogeneous photocatalysis process is electrical energy consumption, hence simple figures-of-merit based on electrical energy consumption can be very useful and informative.41 In the case of low pollutant concentrations in solution, the figures-of-merit “electric energy per order” (EEO) is used in the first order kinetic regime of advanced oxidation processes (AOPs) which is defined by Bolton et al.42 This concept was accepted by the IUPAC as a technical report. Definition of EEO is the number of kilowatt hours of electrical energy needed to reduce the concentration of a pollutant by 1 order of magnitude (90%) in a unit volume of contaminated water.
Nowadays, in most photocatalytic wastewater treatment studies, the catalysts are applied in the form of slurry. However, the separation of catalyst after the reaction in the slurry systems, is a costly step, which adds to the overall running costs of the plant.43 Therefore, for practical application of catalysts, immobilized catalyst systems are preferred in order to prevention the costly separation step.
In this study, Mg and Ag co-impregnated onto TiO2 nanoparticles via impregnation method, and then immobilized on glass plate and used as a fixed-bed system for photocatalytic reduction of Cr(VI) to the less harmful Cr(III). Our main aim of this study was to focusing on minimization of electrical energy consumption and treatment cost during Cr(VI) photoreduction because the most important factor in selecting a waste treatment technology is economics. Thus, the effect of synthesis variables on photocatalytic activity of Mg–Ag/TiO2 nanoparticles was optimized using response surface methodology (RSM). RSM is a widely used statistical technique in process optimizing, and is capable of analyzing the interactions of possible influencing factors and determining the optimum region of the factors level just by using minimum number of designed experiments.44,45 The prepared nanoparticles were characterized by using X-ray diffraction (XRD), transmission electron microscopy (TEM), scanning electron microscopy (SEM), energy dispersive X-ray spectroscopy (EDX), UV-vis diffuse reflectance spectroscopy (DRS), and photoluminescence (PL) techniques.
2. Experimental
2.1. Materials
Titanium dioxide nanoparticles (pure anatase phase, BET surface area 10 m2 g−1), magnesium nitrate (Mg(NO3)2), silver nitrate (AgNO3), and potassium dichromate (K2Cr2O7) were purchased from Merck Co. (Germany).2.2. Preparation of co-impregnated TiO2 nanoparticles
For preparation of magnesium and silver co-impregnated TiO2 nanoparticles, the impregnation method was used according to the following steps. First, 1 g of TiO2 was added to 100 mL deionized water and dispersed for 15 min using a probe sonicator (Bandelin Sonopuls HD 3200, 200 W, 20 kHz), then the required amounts of Mg(NO3)2 and AgNO3 were added to TiO2 suspension. The suspension was stirred for 24 h and then dried in air oven at 80 °C for about 12 h. Then the dried solids calcined at different temperatures for 1 h.2.3. Characterization methods
The prepared nanoparticles were characterized by a Philips X'pert MPD diffractometer using Cu Kα radiation (λ = 0.15478 nm). The average crystallite size of the particles was calculated from the line broadening of corresponding XRD peaks and according to the Scherrer's equation;46
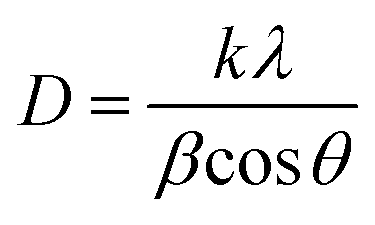 | (1) |
The phase content in the samples was calculated by following equation;47
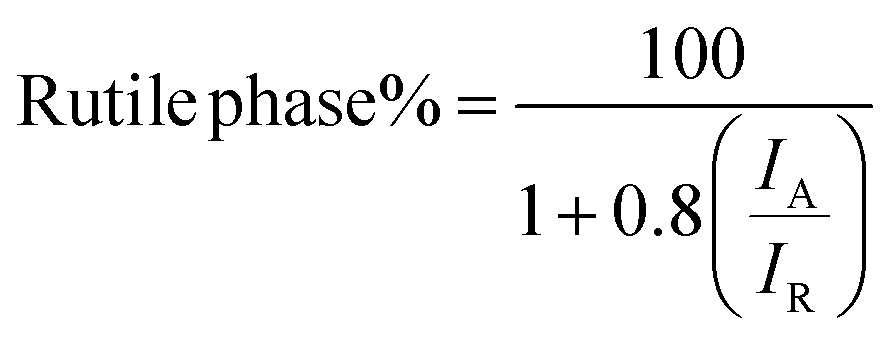 | (2) |
Also, size of the co-impregnated TiO2 sample was obtained by TEM instrument (EM 208 Philips, 100 keV) and the surface morphology of the co-impregnated TiO2 immobilized on glass plate was recorded with KYKY-EM3200 Digital SEM. The SEM was equipped with an EDX system for analyzing the chemical composition of the sample. Photoluminescence emission spectra of the samples was recorded using a Varian Cary-Eclipse luminescence spectrometer (Agilent Technologies) with excitation wavelength at 320 nm.
UV-vis DRS of the samples was obtained using AvaSpec-2048 TEC spectrometer for determination of the optical band gap (Eg) of pure TiO2 and Mg, Ag impregnated and co-impregnated TiO2 nanoparticles. For determination of the Eg eqn (3) was used;
| α(hν) = B(hν − Eg)1/2 | (3) |
2.4. Immobilization of prepared nanoparticles on glass plate
To prepare the immobilized nanoparticles on glass plate (3 cm × 10 cm) heat attachment method was used. In this procedure, a suspension containing 8 g L−1 co-impregnated TiO2 nanoparticles in deionized water was prepared. Then, the prepared suspension was sonicated for 15 min using a probe sonicator in order to improve the dispersion of nanoparticles in deionized water. The effect of ultrasonic irradiation on structural properties of prepared nanoparticles was tested and the results showed that the structural properties of nanoparticles were not affected by ultrasonic irradiation. The sonicated suspension was poured on glass plate and placed in an oven at 100 °C. After drying, the glass plate was fired at 500 °C in a furnace for 1 h and then washed with deionized water for the removal of weakly attached nanoparticles. Catalyst immobilization process was carried out three times to increase the loaded nanoparticles on glass plate. The amount of loaded nanoparticles on the surface of the glass plate was evaluated by the difference in mass of the glass plate before and after immobilization process. Normally, the loaded nanoparticles on the surface was 0.22 mg cm−2.A peel-off test was used on the coating to evaluate the adhesion of nanoparticles immobilized on the glass plate. For this purpose, the absorption spectrum of aqueous colloidal TiO2 in the range of UV (334 nm) has been used.49 First, catalyst immobilized glass plate and 100 mL of distilled water were transferred onto the reactor, and were stirred for 30 min (all conditions were the same as Cr(VI) reduction experiments). Then, samples of water were withdrawn from the reactor after different irradiation times, and the absorbance of the samples were determined by UV-vis spectrophotometer (Rayleigh UV-1600) at 334 nm. Aqueous colloidal TiO2 nanoparticles has a sharp absorption band in the UV range (334 nm), whereas the absorbance of the immobilized sample were near to zero for all irradiation times. This result confirmed that prepared nanoparticles were strictly immobilized on the glass plate.
2.5. Photocatalytic reduction experiments
Photocatalytic reduction processes were carried out at room temperature in a batch quartz reactor. Artificial irradiation was provided by 15 W (UV-C) mercury lamp (Philips, Holland) emitting around 254 nm, positioned in top of the batch quartz reactor. In each run, a glass plate loaded with co-impregnated TiO2 nanoparticles was inserted in quartz reactor, then 100 mL of the prepared solution with Cr(VI) concentration of 5 mg L−1 was transferred onto the reactor and was stirred for 30 min to reach the adsorption equilibration in darkness before irradiation. The photocatalytic reaction was initiated with turning on the light source. At given irradiation time intervals, the samples (5 mL) were taken out, and then Cr(VI) concentration analyzed by UV-vis spectrophotometer at λmax = 350 nm.2.6. Electrical energy determination
The electric energy per order (kW h per m3 per order) required to the photocatalytic reduction of Cr(VI) was calculated from the following equations:42
 | (4) |
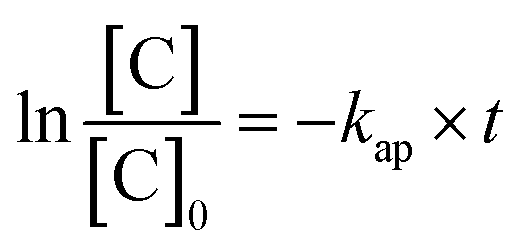 | (5) |
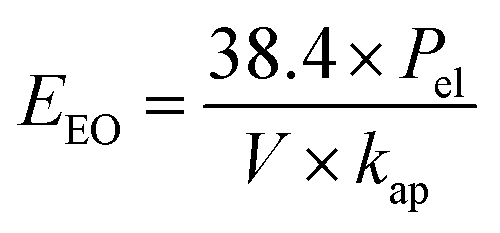 | (6) |
2.7. Experimental design
Central composite design (CCD) that is the most common form of RSM technique, was used for the optimization of Mg, Ag co-impregnated TiO2 preparation conditions. In order to evaluate the effect of synthesis variables on photocatalytic activity of co-impregnated TiO2 nanoparticles, three key factors were selected: Mg content, Ag content, and calcination temperature and the photocatalytic reduction of Cr(VI) was selected as the response. A total of 20 experiments runs were performed in this work with six replications at the center point. Computational analysis of the experimental data was supported by the Design-Expert (version 8) software. For statistical calculations, the three chosen synthesis variables were converted to dimensionless ones (x1, x2, x3), with the coded values at levels: −2, −1, 0, +1, +2. The experimental ranges and the levels of the synthesis variables are presented in Table S1 of ESI.† Model analysis results are provided in Text S1 of ESI.†3. Results and discussion
The details of the designed experiments along with experimental results and predicted values for photocatalytic reduction efficiencies of Cr(VI) are presented in Table 1. Contour plots help in identification of the type of interactions between synthesis conditions and the three-dimensional surface plots are useful approach in revealing of synthesis conditions. The contour and response surface plots for synthesis variables, while one variable kept at its respective zero level and the others varying within the experimental ranges, are obtained to evaluate the interactive relationships between the selected factors and photocatalytic reduction rate of Cr(VI) as the response. Fig. 1a shows the effect of the magnesium and silver weight ratio on the photocatalytic activity of co-impregnated TiO2 nanoparticles, while other variable was kept at its respective zero level (calcination temperature of 450 °C). As can be seen from Table 1, photocatalytic reduction rate of Cr(VI) increased from 32.59% to 68.01% with increasing the Ag content from 0 to 2 wt%. The Ag nanoparticles onto TiO2 act as an efficient electron scavenger, through indirectly modification of the interfacial charge transfer processes (ICTP). When Ag make contact with TiO2, due to the formation of Schottky barrier in Ag and TiO2 contact region, and because the Fermi level of Ag is lower than that of TiO2 conduction band, electrons will transfer from the conduction band of TiO2 to Ag.50–52 The electrons transferred to the Ag cluster react with adsorbed oxygen molecules to form superoxide anions and free valence band holes of TiO2 react with adsorbed water molecules and hydroxide ions to produce hydroxyl radicals as reactive species in the degradation process.53,54 Finally, this process provides the separation of electron–hole pairs and results in the improvement of the photocatalytic activity of the TiO2 nanoparticles. Moreover, Ag nanoparticles have plasmonic resonance in the optical range of UV and near-UV-visible light, which increases significantly their photon absorption efficiency. Silver nanoparticles can harvest and transfer more light energy to the TiO2 catalyst at the energies above and below the band edge through resonant energy transfer. This phenomenon leads to generating more electron–hole pairs in the TiO2 catalyst and resulting in enhancement of photocatalytic activity.55–58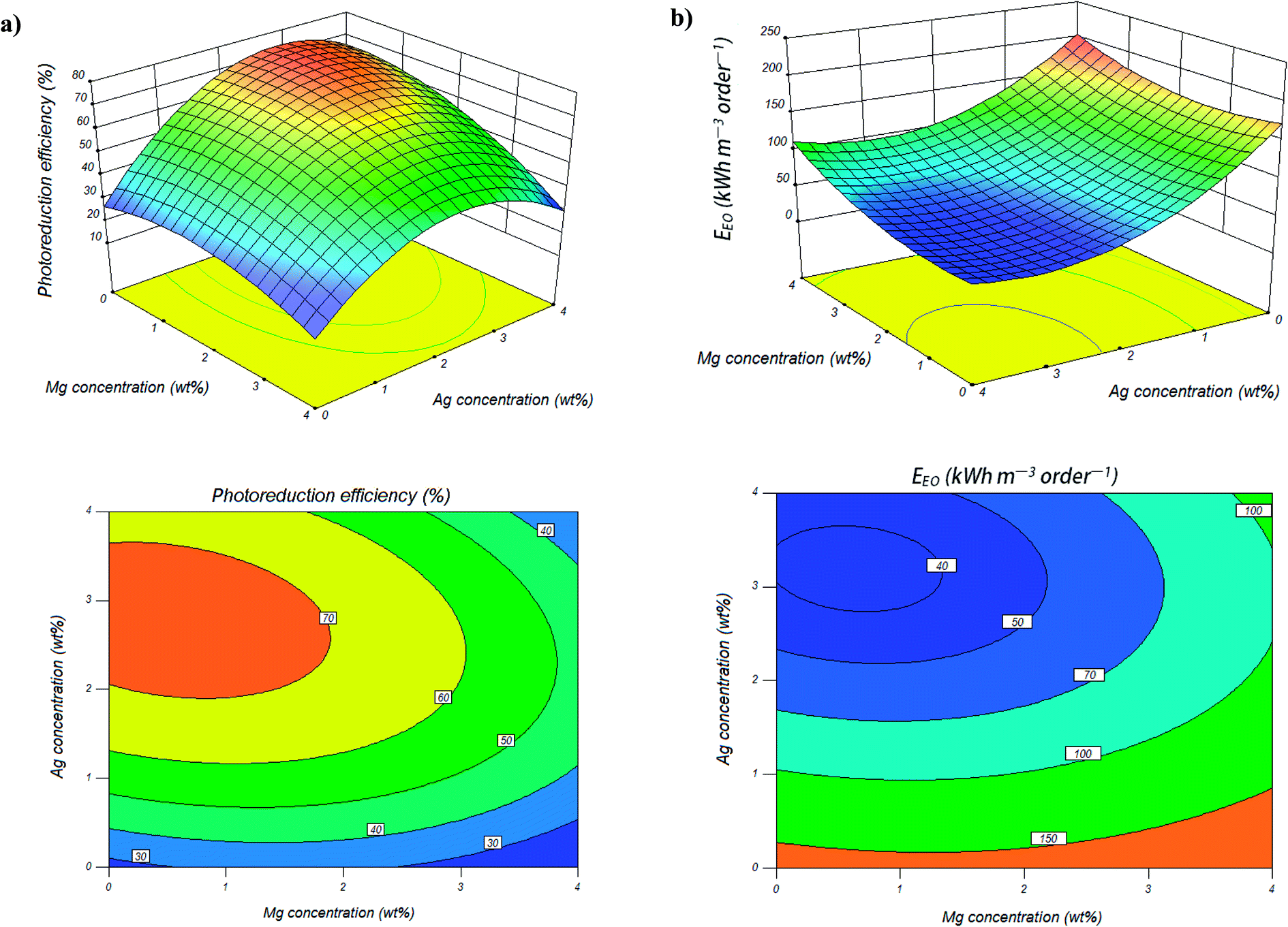 | ||
| Fig. 1 The response surface and contour plots of photocatalytic reduction efficiency of Cr(VI) (a) and electrical energy consumption (b), as a function of Mg and Ag contents (calcination temperature of 450 °C). | ||
| Run | Mg concentration (wt%) | Ag concentration (wt%) | Calcination temperature (°C) | Photocatalytic reduction (%) | |
|---|---|---|---|---|---|
| Experimental | Predicted | ||||
| 1 | 2 | 2 | 450 | 68.01 | 67.47 |
| 2 | 2 | 0 | 450 | 32.59 | 30.81 |
| 3 | 4 | 2 | 450 | 43.11 | 46.59 |
| 4 | 2 | 2 | 450 | 67.09 | 67.47 |
| 5 | 3.2 | 0.8 | 550 | 36.63 | 35.59 |
| 6 | 0.8 | 0.8 | 350 | 28.34 | 27.76 |
| 7 | 2 | 2 | 450 | 67.29 | 67.47 |
| 8 | 3.2 | 0.8 | 350 | 31.47 | 32.31 |
| 9 | 2 | 2 | 450 | 68.37 | 67.47 |
| 10 | 2 | 2 | 282 | 29.62 | 32.55 |
| 11 | 2 | 2 | 450 | 68.01 | 67.47 |
| 12 | 2 | 4 | 450 | 45.97 | 47.02 |
| 13 | 3.2 | 3.2 | 550 | 44.74 | 38.75 |
| 14 | 2 | 2 | 618 | 41.3 | 47.65 |
| 15 | 0.8 | 3.2 | 550 | 77.85 | 73.44 |
| 16 | 0.8 | 3.2 | 350 | 61.28 | 55.76 |
| 17 | 0.8 | 0.8 | 550 | 59.22 | 57.65 |
| 18 | 3.2 | 3.2 | 350 | 55.69 | 50.69 |
| 19 | 2 | 2 | 450 | 67.65 | 67.47 |
| 20 | 0 | 2 | 450 | 63.61 | 69.41 |
On the other hand, as can be seen from Table 1, photocatalytic reduction rate of Cr(VI) increased from 63.61% to about 68.01% with increasing the Mg content from 0 to 2 wt%. The positive effect of Mg on the photocatalytic activity, can be explained by Mg ions ability to trap electrons at conduction band of TiO2 and extending the lifetime of the photogenerated electron–hole pairs. The trapped electrons can be subsequently scavenged by molecular oxygen, which is adsorbed on the surface of TiO2, and generate reactive species such as superoxide radicals, hydroxyl radicals, and hydrogen peroxide, through the following reactions:59
| Mg2+ − TiO2 + e−CB → Mg+ − TiO2 (unstable) | (7) |
| Mg+ − TiO2 + O2 → Mg2+ − TiO2 + O2˙− | (8) |
| O2˙− + H+ → HO2˙ | (9) |
| HO2˙ + O2˙− + H+ → H2O2 + O2 | (10) |
| H2O2 + e−CB → ˙OH + OH− | (11) |
| H2O2 + O2˙− → ˙OH + OH− + O2 | (12) |
| OH− + hVB+ → ˙OH | (13) |
As can be seen from the response surface and contour plots (Fig. 1a), further increasing the Mg and Ag content can lead to a decrease in photocatalytic activity of TiO2 nanoparticles. This can be the result of following reasons: one explanation is that with increasing the number and size of metal clusters they become recombination centers for electron–hole pairs, and another explanation is that more numbers of negatively charged metal sites onto TiO2 system, screen the TiO2 surface from UV light absorption, and results lead to decrease in photocatalytic activity of the TiO2 nanoparticles.21,51,60,61 The contour plots (Fig. 1a) show that the optimum region for highest photocatalytic reduction efficiency (≥80%) is the Mg content less than 1.5 wt% and Ag content in range of 2–3.5 wt%, when calcination temperature is kept at constant 450 °C.
Electrical energy required to the photocatalytic reduction of Cr(VI) in the presence of co-impregnated TiO2 nanoparticles with different Mg and Ag metals content calculated from the eqn (6) and showed in Fig. 1b. Results showed that less energy was consumed during the photocatalytic reduction of Cr(VI) in the presence of Mg, Ag co-impregnated TiO2 nanoparticles in comparison to mono-impregnated TiO2 nanoparticles. Also, electrical energy required to the photocatalytic reduction process was also affected by Mg and Ag metals content. The contour plots (Fig. 1b) show that the optimum region for minimum electrical energy consumption (≤40 kW h per m3 per order) during Cr(VI) reduction is when the Mg content less than 1 wt% and Ag content in range of 3–3.5 wt%, which this result is in agreement with the high photocatalytic activity of Mg, Ag co-impregnated TiO2 nanoparticles showed in the contour plots (Fig. 1a).
On the other hand, photocatalytic reduction efficiency of Cr(VI) was also affected by calcination temperature. Fig. 2a shows the response surface and contour plots of photocatalytic reduction efficiency of Cr(VI) as a function of calcination temperature and Mg content, while Ag content is kept at its respective zero level. As it is obvious from Fig. 2a and Table 1, a slightly improvement in photocatalytic activity of Mg, Ag co-impregnated TiO2 nanoparticles obtained with increasing of calcination temperature from 282 to 450 °C. However, further increase of the calcination temperature from 450 to 618 °C can leads to a significant decrease in photocatalytic reduction rate of Cr(VI). XRD patterns of calcined samples at different temperatures in Fig. 3 show that the calcination temperature is very effective parameter in changing the phase content of co-impregnated TiO2 nanoparticles. XRD patterns reveal that the co-impregnated TiO2 nanoparticles calcined at 282 °C exists solely as anatase phase, whereas samples calcined at 450 and 618 °C have mixed anatase/rutile phases. The presence of small quantities of rutile phase in adjacent to anatase phase, act as a structural defect or impurity and promotes the electrons transport from the conduction band of anatase to that of rutile.62–64 This electrons transport can be effective in decreasing recombination rate of electron–hole pairs and enhancing photocatalytic activity. On the other hand, photocatalytic activity is in relation with crystallite size. The average crystallite size of co-impregnated TiO2 nanoparticles calcined at different temperatures were calculated from the eqn (1) using reflections of anatase at 25.3° and rutile at 27.4°. The average crystallite sizes are estimated to be 36, 41, and 54 nm for samples calcined at 282, 450, and 618 °C, respectively, which means that calcination of nanoparticles at higher temperature, results in larger particle size. The photocatalytic activity decreases with further increasing in calcination temperature can be attributed to the transformation of anatase to the rutile phase, increase in crystallite size of particles, and agglomeration of nanoparticles.62,65,66
 | ||
| Fig. 2 The response surface and contour plots of photocatalytic reduction efficiency of Cr(VI) (a) and electrical energy consumption (b), as a function of Mg content and calcination temperature (Ag content of 2 wt%). | ||
 | ||
| Fig. 3 XRD patterns of Mg, Ag co-impregnated TiO2 (2–2 wt%) nanoparticles calcined at different temperatures. | ||
Results of electrical energy consumption (Fig. 2b) show that the electrical energy required to the photocatalytic reduction of Cr(VI) was also affected by calcination temperature. The contour plots (Fig. 2b) show that the optimum region for minimum electrical energy consumption (≤50 kW h per m3 per order) during Cr(VI) reduction is when the Mg content less than 1.5 wt% and calcination temperature in range of 470–550 °C, which this result is in agreement with the high photocatalytic activity of Mg, Ag co-impregnated TiO2 nanoparticles showed in the contour plots (Fig. 2a).
The main aim in this study was to achieve optimum synthesis conditions for the preparation of photocatalyst with highest photocatalytic activity to reduce electrical energy consumption. Design Expert as a response optimizer software was used for the optimization of synthesis conditions in the selected range that the Mg content, Ag content, and calcination temperature are in the range of 0–4 wt%, 0–4 wt%, and 282–618 °C, respectively. The optimal values of the synthesis variables for the maximum photocatalytic reduction efficiency of Cr(VI) (84.44%) and minimum electrical energy consumption (30.31 kW h per m3 per order), were 0.82 wt%, 2.6 wt%, and 495 °C for Mg content, Ag content, and calcination temperature, respectively. As consequent, experimental design strategy can be a successful technique to determine the optimum synthesis conditions to reduce electrical energy consumption and can be an adequate modeling to predict the photocatalytic reduction rate of Cr(VI). Fig. 4 shows the plot of Cr(VI) concentration as a function of irradiation time during photocatalytic reduction in the presence of Mg, Ag co-impregnated TiO2 prepared under optimized conditions.
 | ||
| Fig. 4 Change in Cr(VI) concentration as a function of irradiation time during photocatalytic reduction in the presence of Mg, Ag co-impregnated TiO2 (0.82–2.6 wt%) nanoparticles calcined at 495 °C. | ||
The EEO amount for pure TiO2 (232.1 kW h per m3 per order), Mg-impregnated TiO2 (152.03 kW h per m3 per order), and Ag-impregnated TiO2 (71.42 kW h per m3 per order) is about 8, 5, and 2 times more than that of optimized Mg, Ag co-impregnated TiO2 nanoparticles, respectively. It is useful to relate the values of electrical energy found in this work to the operation costs. By considering 0.036 US $ per kW per h as the cost of electricity in Iran, the contribution to operation costs of Cr(VI) reduction from electrical energy will be 8.35, 5.47, 2.57, and 1.09 US $ per m3, for pure TiO2, Mg-impregnated TiO2, Ag-impregnated TiO2, and Mg, Ag co-impregnated TiO2 nanoparticles, respectively. Thus, the Mg, Ag co-impregnated TiO2 with optimized of synthesis variables is more economical than other photocatalysts, due to the significant reduction in energy required.
The reusability of fixed-bed catalytic system was evaluated in the photocatalytic reduction of Cr(VI). After reaction cessation, the immobilized Mg, Ag co-impregnated TiO2 nanoparticles was removed from photoreactor and washed with water several times. After drying at 100 °C during 5 h, the immobilized nanoparticles were reused in photocatalytic reduction of fresh Cr(VI) solution. The photocatalytic reduction efficiency of Cr(VI) after first, second, third, and fourth reuse cycle were 34.7, 27.6, 25.1, and 21.9%, respectively. It is obvious that the photocatalytic activity of fixed-bed system deteriorated with the increase in the reuse cycles number. Similar results have also been reported by Li et al.67 and Zhang et al.68 for the photocatalytic reduction of Cr(VI) in the presence of SnS2/TiO2. The hydrolysis-precipitation of generated Cr(III) ions, led to the formation of Cr(OH)3 species on the surface of catalyst, which these species likely occupy some photocatalytic active sites of catalyst and resulting to decrease in the photocatalytic activity of catalyst during its reuse.67,68
The mean particle size of the Mg, Ag co-impregnated TiO2 sample prepared under optimized conditions was further investigated with TEM analysis. From TEM image in Fig. 5a the mean particle size of optimized Mg, Ag co-impregnated TiO2 nanoparticles are estimated to be about 50 nm, which this result is in agreement with the average crystallite size calculated from the XRD pattern. The composition of the co-impregnated TiO2 sample was investigated with EDX analysis at the microscopic level. The results from EDX spectrum (Fig. 5b) clearly confirm the existence of Mg and Ag on the co-impregnated TiO2 nanoparticles.
 | ||
| Fig. 5 TEM image (a) and EDX spectrum (b) of the Mg, Ag co-impregnated TiO2 (0.82–2.6 wt%) nanoparticles calcined at 495 °C. | ||
Fig. 6a shows the SEM micrograph of Mg, Ag co-impregnated TiO2 (0.82–2.6 wt%) nanoparticles calcined at 495 °C. This image shows particles with uniform distribution, spherical morphology, and slight agglomeration. Fig. 6b shows SEM micrograph from the cross section of the fixed-bed. It indicates thickness of ∼3.4 μm for immobilized nanoparticles on the glass plate. SEM micrograph from the surface of the immobilized nanoparticles on the glass plate is presented in Fig. S2 of ESI.† It is obvious that during the immobilization stages of nanoparticles on glass plate the agglomeration of nanoparticles was not happened.
 | ||
| Fig. 6 SEM micrographs of Mg, Ag co-impregnated TiO2 (0.82–2.6 wt%) nanoparticles calcined at 495 °C (a) and immobilized nanoparticles on the glass plate, picture from the cross section (b). | ||
To study the effect of metals impregnation on the optical absorption properties of TiO2 nanoparticles, DRS analysis has been carried out. The reflectance spectrum showed the significantly red shifted absorption wavelengths for Mg/TiO2, Ag/TiO2, and co-impregnated Mg–Ag/TiO2. The values of band gap energy (Eg) are calculated from Fig. 7 by extrapolation of the linear part of the spectra to the energy axis. The Eg values for pure TiO2, Mg-impregnated TiO2, Ag-impregnated TiO2, and Mg, Ag co-impregnated TiO2 nanoparticles are 3.26, 3.13, 3.04, and 2.99 eV. Results indicate that impregnation of Mg and Ag metals onto TiO2 nanoparticles decreased optical band gap energy, whereas decrease in Eg value for co-impregnated TiO2 nanoparticles is higher in comparison to the individual impregnated nanoparticles.
 | ||
| Fig. 7 Plot of (αhν)2 versus hν for pure TiO2, Mg-impregnated TiO2 (2 wt%), Ag-impregnated TiO2 (2 wt%), and Mg, Ag co-impregnated TiO2 (0.82–2.6 wt%) nanoparticles. | ||
The PL emission in semiconductors arises from the recombination of free charge carriers, therefore the PL spectra used to study the transfer, migration, and recombination processes of the photogenerated electron–hole pairs.69,70 Fig. 8 shows the PL spectra of the pure TiO2, mono-impregnated, and co-impregnated TiO2 nanoparticles excited by 320 nm. From this figure, it can be observed that there is a significant decrease in the intensity of PL spectra of co-impregnated TiO2 compared to that of the pure and mono-impregnated TiO2. Weaker intensity of the peak suggests that the Mg and Ag co-impregnated on the surface of TiO2 with could effectively inhibit the recombination probability of photogenerated electrons and holes, due to the separately charge transfer between the metals and TiO2. The considerable PL quenching is observed in the co-impregnated TiO2 nanoparticles with optimized metals content (Mg content of 0.82 wt% and Ag content of 2.6 wt%).
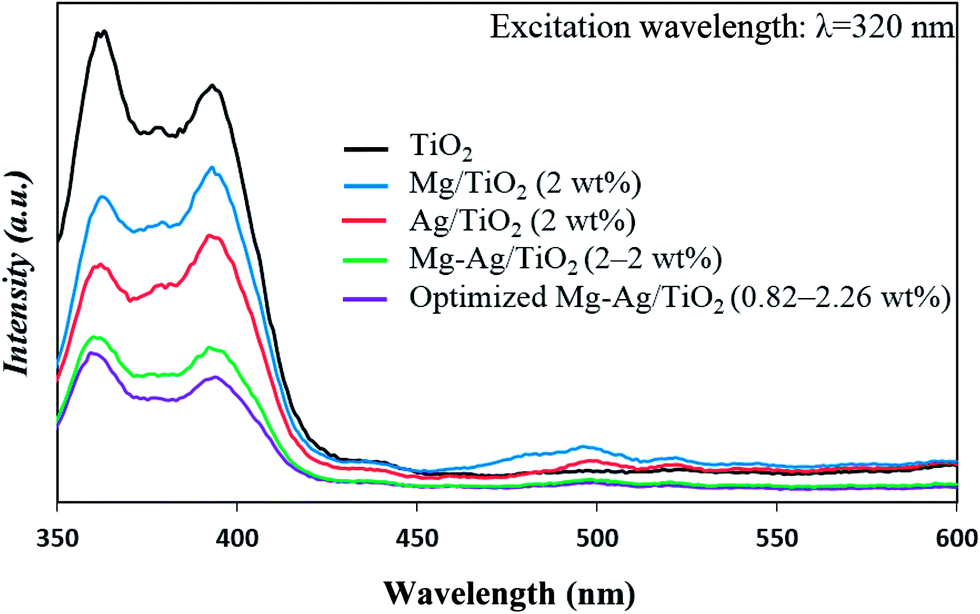 | ||
| Fig. 8 PL spectra of pure TiO2, Mg-impregnated TiO2 (2 wt%), Ag-impregnated TiO2 (2 wt%), and Mg, Ag co-impregnated TiO2 nanoparticles. | ||
4. Conclusions
Mg, Ag co-impregnated TiO2 nanoparticles were prepared via impregnation method and employed in the photocatalytic reduction of Cr(VI) to the less harmful Cr(III) in a fixed-bed catalytic system. TEM analysis indicated a mean particle size of 50 nm for optimized co-impregnated TiO2 nanoparticles. DRS results indicated a considerable decrease in Eg value for co-impregnated nanoparticles in comparison with pure and mono-impregnated TiO2 nanoparticles. RSM technique was successfully used for optimizing photocatalytic activity of co-impregnated TiO2 nanoparticles with focusing on minimization of electrical energy consumption. At the optimized preparation conditions (0.82 wt% Mg, 2.6 wt% Ag, and 495 °C calcination temperature) the maximum photocatalytic reduction efficiency (84.44%) and minimum electrical energy consumption (30.31 kW h per m3 per order) were achieved for co-impregnated TiO2 nanoparticles, whereas at the same conditions, EEO amounts using pure TiO2, Mg-impregnated TiO2 and Ag-impregnated TiO2 samples were 232.1, 152.03, and 71.42 kW h per m3 per order, respectively. PL results indicated a considerable quenching in the co-impregnated TiO2 with optimized metals content that suggests the co-impregnation of metals onto TiO2 could effectively inhibit the recombination probability of photogenerated electrons–holes pairs. Co-impregnation of Mg and Ag by its ability to separately trap photogenerated electrons provide a synergistic effect in the enhancement of the TiO2 nanoparticles photocatalytic activity and effectively minimization of the electrical energy consumption and treatment cost in photocatalytic reduction of Cr(VI).Acknowledgements
The authors would like to thank University of Tehran for financial support of this work.References
- S. Velazquez-Peña, I. Linares-Hernández, V. Martínez-Miranda, C. Barrera-Díaz, V. Lugo-Lugo and B. Bilyeu, Sep. Sci. Technol., 2013, 48, 2900–2909 CrossRef.
- S. Rengaraj, K. H. Yeon and S. H. Moon, J. Hazard. Mater., 2001, 87, 273–287 CrossRef CAS.
- G. Q. Chen, W. J. Zhang, G. M. Zeng, J. H. Huang, L. Wang and G. L. Shen, J. Hazard. Mater., 2011, 186, 2138–2143 CrossRef CAS PubMed.
- M. Costa, Toxicol. Appl. Pharmacol., 2003, 188, 1–5 CrossRef CAS.
- M. K. Aroua, F. M. Zuki and N. M. Sulaiman, J. Hazard. Mater., 2007, 147, 752–758 CrossRef CAS PubMed.
- F. D. Natale, A. Lancia, A. Molino and D. Musmarra, J. Hazard. Mater., 2007, 145, 381–390 CrossRef PubMed.
- T. Karthikeyan, S. Rajgopal and L. R. Miranda, J. Hazard. Mater., 2005, 124, 192–199 CrossRef CAS PubMed.
- N. Kongsricharoern and C. Polprasert, Water Sci. Technol., 1996, 34, 109–116 CrossRef CAS.
- P. Lakshmipathiraj, G. Bhaskar Raju, M. Raviatul Basariya, S. Parvathy and S. Prabhakar, Sep. Purif. Technol., 2008, 60, 96–102 CrossRef CAS PubMed.
- J. Yoon, G. Amy, J. Chung, J. Sohn and Y. Yoon, Chemosphere, 2009, 77, 228–235 CrossRef CAS PubMed.
- S. L. Lo and S. F. Shiue, Water Res., 1998, 32, 174–178 CrossRef CAS.
- Y. Xing, X. Chen and D. Wang, Environ. Sci. Technol., 2007, 41, 1439–1443 CrossRef CAS.
- F. Gode and E. Pehlivan, J. Hazard. Mater., 2005, 119, 175–182 CrossRef CAS PubMed.
- S. D. Huang, C. F. Fann and H. S. Hsieh, J. Colloid Interface Sci., 1982, 89, 504–513 CrossRef CAS.
- S. D. Huang and D. J. Wilson, Sep. Purif. Technol., 1984, 19, 603–611 CAS.
- L. B. Khalil, W. E. Mourad and M. W. Rophael, Appl. Catal., B, 1998, 17, 267–273 CrossRef CAS.
- N. Daneshvar, D. Salari and S. Aber, J. Hazard. Mater., 2002, 94, 49–61 CrossRef CAS.
- S. Rengaraj, S. Venkataraj, J. W. Yeon, Y. Kim, X. Z. Li and G. K. H. Pang, Appl. Catal., B, 2007, 77, 157–165 CrossRef CAS PubMed.
- P. Mohapatra, S. K. Samantaray and K. Parida, J. Photochem. Photobiol., A, 2005, 170, 189–194 CrossRef CAS PubMed.
- L. Yang, Y. Xiao, S. Liu, Y. Li, Q. Cai, S. Luo and G. Zeng, Appl. Catal., B, 2010, 94, 142–149 CrossRef CAS PubMed.
- M. A. Behnajady and H. Eskandarloo, J. Nanosci. Nanotechnol., 2013, 13, 548–553 CrossRef CAS PubMed.
- J. Zhang, Q. Xu, Z. Feng, M. Li and C. Li, Angew. Chem., Int. Ed., 2008, 47, 1766–1769 CrossRef CAS PubMed.
- M. A. Behnajady, N. Modirshahla, M. Shokri, H. Elham and A. Zeininezhad, J. Environ. Sci. Health, Part A, 2008, 43, 460–467 CrossRef CAS PubMed.
- M. A. Behnajady, N. Mansoriieh, N. Modirshahla and M. Shokri, Environ. Technol., 2012, 33, 265–271 CrossRef CAS.
- L. Zhong, F. Haghighat, P. Blondeau and J. Kozinski, Build. Environ., 2010, 45, 2689–2697 CrossRef PubMed.
- S. Deng and R. Bai, Water Res., 2004, 38, 2424–2432 CrossRef CAS PubMed.
- V. Sarin and K. K. Pant, Bioresour. Technol., 2006, 97, 15–20 CrossRef CAS PubMed.
- B. Xin, L. Jing, Z. Ren, B. Wang and H. Fu, J. Phys. Chem. B, 2005, 109, 2805–2809 CrossRef CAS PubMed.
- Y. Zhang, N. Zhang, Z. R. Tang and Y. J. Xu, ACS Sustainable Chem. Eng., 2013, 1, 1258–1266 CrossRef CAS.
- W. Chen, Z. Fan and Z. Lai, J. Mater. Chem. A, 2013, 1, 13862–13868 CAS.
- Z. Li, Y. Hu and Y. Sun, Chem. Commun., 2014, 50, 1411–1413 RSC.
- G. Liu, L. Wang, H. G. Yang, H. M. Cheng and G. Q. M. Lu, J. Mater. Chem., 2010, 20, 831–843 RSC.
- J. W. Shi, J. T. Zheng, Y. Hu and Y. C. Zhao, Mater. Chem. Phys., 2007, 106, 247–249 CrossRef CAS PubMed.
- A. Naldoni, M. D'Arienzo, M. Altomare, M. Marelli, R. Scotti, F. Morazzoni, E. Selli and V. Dal Santo, Appl. Catal., B, 2013, 130–131, 239–248 CrossRef CAS PubMed.
- Z. Liu, J. Chen, Y. Zhang, L. Wu and X. Li, Mater. Chem. Phys., 2011, 128, 1–5 CrossRef CAS PubMed.
- K. Kočí, K. Matějů, L. Obalová, S. Krejčíková, Z. Lacný, D. Plachá, L. Čapek, A. Hospodková and O. Šolcová, Appl. Catal., B, 2010, 96, 239–244 CrossRef PubMed.
- V. Diesen, C. W. Dunnill, E. Österberg, I. P. Parkin and M. Jonsson, Dalton Trans., 2014, 43, 344–351 RSC.
- K. Kakiage, T. Tokutome, S. Iwamoto, T. Kyomen and M. Hanaya, Chem. Commun., 2013, 49, 179–180 RSC.
- M. Manzanares, C. Fàbrega, J. OriolOssó, L. F. Vega, T. Andreu and J. R. Morante, Appl. Catal., B, 2014, 150, 57–62 CrossRef PubMed.
- R. Mohamed, Z. Khusaimi, A. N. Afaah, A. Aadila, M. H. Mamat and M. Rusop, J. Nano Res., 2014, 26, 33–38 CrossRef CAS.
- M. B. Kasiri and A. R. Khataee, Environ. Technol., 2012, 33, 1417–1425 CrossRef CAS.
- J. R. Bolton, K. G. Bircher, W. Tumas and C. A. Tolman, Pure Appl. Chem., 2001, 73, 627–637 CrossRef CAS.
- M. Zulfakar and H. Nazirah, J. Appl. Sci., 2011, 11, 2320–2326 CrossRef CAS.
- G. Wang, Y. Mu and H. Q. Yu, Biochem. Eng. J., 2005, 23, 175–184 CrossRef CAS PubMed.
- J. Chen, G. Li, Y. Huang, H. Zhang, H. Zhao and T. An, Appl. Catal., B, 2012, 123–124, 69–77 CrossRef CAS PubMed.
- A. L. Patterson, Phys. Rev., 1939, 56, 978–982 CrossRef CAS.
- R. A. Spurr and H. Myers, Anal. Chem., 1957, 29, 760–762 CrossRef CAS.
- A. M. Abdul-Kader, Philos. Mag. Lett., 2009, 89, 162–169 CrossRef CAS.
- J. A. Byrne, B. R. Eggins, N. M. D. Brown, B. McKinney and M. Rouse, Appl. Catal., B, 1998, 17, 25–36 CrossRef CAS.
- M. S. Lee, S. S. Hong and M. Mohseni, J. Mol. Catal. A: Chem., 2005, 242, 135–140 CrossRef CAS PubMed.
- M. A. Behnajady and H. Eskandarloo, Chem. Eng. J., 2013, 228, 1207–1213 CrossRef CAS PubMed.
- M. K. Seery, R. George, P. Floris and S. C. Pillai, J. Photochem. Photobiol., A, 2007, 189, 258–263 CrossRef CAS PubMed.
- M. El-Kemary, Y. Abdel-Moneam, M. Madkour and I. El-Mehasseb, J. Lumin., 2011, 131, 570–576 CrossRef CAS PubMed.
- L. Ren, Y. P. Zeng and D. Jiang, Chem. Commun., 2009, 10, 645–649 CAS.
- Y. Qu and X. Duan, Chem. Soc. Rev., 2013, 42, 2568–2580 RSC.
- A. Kubacka, M. L. Cerrada, C. Serrano, M. Fernández-García, M. Ferrer and M. Fernández-Garcia, J. Phys. Chem. C, 2009, 113, 9182–9190 CAS.
- Á. Veres, T. Rica, L. Janovák, M. Dömök, N. Buzas, V. Zöllmer, T. Seemann, A. Richardt and I. Dékány, Catal. Today, 2012, 181, 156–162 CrossRef PubMed.
- J. Li, S. K. Cushing, J. Bright, F. Meng, T. R. Senty, P. Zheng, A. D. Bristow and N. Wu, ACS Catal., 2012, 3, 47–51 CrossRef.
- B. K. Avasarala, S. R. Tirukkovalluri and S. Bojja, J. Hazard. Mater., 2011, 186, 1234–1240 CrossRef CAS PubMed.
- H. M. Coleman, K. Chiang and R. Amal, Chem. Eng. J., 2005, 113, 65–72 CrossRef CAS PubMed.
- T. T. Y. Tan, C. K. Yip, D. Beydoun and R. Amal, Chem. Eng. J., 2003, 95, 179–186 CrossRef CAS.
- M. A. Behnajady, H. Eskandarloo, N. Modirshahla and M. Shokri, Desalination, 2011, 278, 10–17 CrossRef CAS PubMed.
- N.-G. Park, G. Schlichthörl, J. van de Lagemaat, H. M. Cheong, A. Mascarenhas and A. J. Frank, J. Phys. Chem. B, 1999, 103, 3308–3314 CrossRef CAS.
- N. G. Park, J. van de Lagemaat and A. J. Frank, J. Phys. Chem. B, 2000, 104, 8989–8994 CrossRef CAS.
- M. A. Behnajady, H. Eskandarloo, N. Modirshahla and M. Shokri, Photochem. Photobiol., 2011, 87, 1002–1008 CrossRef CAS PubMed.
- H. I. Hsiang and S. C. Lin, Ceram. Int., 2008, 34, 557–561 CrossRef CAS PubMed.
- J. Li, T. Wang and X. Du, Sep. Purif. Technol., 2012, 101, 11–17 CrossRef CAS PubMed.
- Y. C. Zhang, J. Li and H. Y. Xu, Appl. Catal., B, 2012, 123–124, 18–26 CrossRef CAS PubMed.
- J. Ren, W. Wang, S. Sun, L. Zhang and J. Chang, Appl. Catal., B, 2009, 92, 50–55 CrossRef CAS PubMed.
- J. Tian, Y. Sang, Z. Zhao, W. Zhou, D. Wang, X. Kang, H. Liu, J. Wang, S. Chen, H. Cai and H. Huang, Small, 2013, 9, 3864–3872 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra03418j |
| This journal is © The Royal Society of Chemistry 2014 |