Two alternative approaches to the Diels–Alder polymerization of tung oil
Talita M. Lacerda*ab,
Antonio J. F. Carvalhob and
Alessandro Gandiniab
aEscola de Engenharia de São Carlos, Universidade de São Paulo, 13566-590, São Carlos, SP, Brazil. E-mail: talita@iqsc.usp.br; Fax: +55 16 33739590; Tel: +55 16 33738679
bInstituto de Química de São Carlos, Universidade de São Paulo, 13560-970, São Carlos, SP, Brazil
First published on 9th June 2014
Abstract
Novel linear and crosslinked polymers from non-modified and modified tung oil are reported. These materials are based on the exploitation of the Diels–Alder reaction using, on the one hand, the dienic character of the three conjugated double bonds of tung oil and their susceptibility to react with a dienophile, and, on the other hand, the bulk reaction of furfuryl amine at the ester moieties of tung oil to produce three fatty acid furan amides, still bearing the three conjugated double bonds, and their linear polymerization with bismaleimides.
Introduction
The realm of polymers from renewable resources has been experiencing an outstanding progress in recent years, both qualitatively and quantitatively,1 and the exploitation of plant oils2 and furan derivatives3 are among the most active areas of investigation, either in their respective fields or in studies involving the conjunct use of both types of precursors.4 Vegetable oils have been used as such, or after suitable chemical modifications, and the variety of macromolecular materials reported in the last couple of decades have called upon a rich set of alternative polymerization mechanisms and cover an impressive range of structures, properties and promising applications.2 The same applies to polymers based on furan monomers and furan chemistry, with recent original contributions based on Diels–Alder (DA) polycondensations3,5 and on the use of 2,5-furandicarboxylic acid for the synthesis of polyesters3,6 and polyamides.7 The latter context is associated with the booming interest in the search for optimal conditions associated with the preparation of hydroxymethylfurfural from sugars and polysaccharides.8Tung oil (TO), easily obtained from the seeds of tung tree (Vernicia fordii) nuts, is a relatively cheap commodity which has been used for millennia as a source of siccative coatings, incorporates as its main constituent (∼85%) a peculiar triglyceride structure in which each chain bears three conjugated unsaturations corresponding to α-eleostearic acid.
Although its use as a precursor to novel polymers has been much less important to date than that of such oils as soybean, castor and linseed, a number of studies have been devoted in recent years to new approaches toward the polymerization of pristine TO or of monomers derived from its chemical modifications. These include materials based on (i) TO, styrene9 and divinylbenzene,10 (ii) the cationic polymerization of the oil,11 (iii) monomers derived from the introduction of carboxylic groups onto the oil structure using its DA reaction with maleic anhydride,9,12 (iv) TO-based polyurethanes,13 and (v) the interaction of the oil with a bismaleimide.14 The latter report is in fact the only publication dealing with the direct DA polymerization of TO, used as a dienic monomer, but its interest is limited by the fact that, on the one hand, very drastic temperature conditions were adopted to promote the couplings, opening the way to side reactions and, on the other hand, the authors limited their study to the use of a single bismaleimide and only characterized a single crosslinked material. We report here a more systematic approach to the DA polymerization of TO under milder conditions, using a selection of bismaleimides in order to obtain products with different properties, notably in terms of glass transition temperatures.
In another vein, we recently showed that furfuryl amine readily reacts with epoxidized triglycerides through two parallel mechanisms, one based on the aminolysis of their ester groups and the other on the classical oxirane ring opening by the primary amino function, but without the use of a catalyst or a solvent.15 This led to furan/oil-based monomers incorporating two or three heterocycles, which were then polymerized using an aromatic bismaleimide to generate linear and branched macromolecules that were thoroughly characterized. Here, we took advantage of the aminolysis reaction to convert TO into three macromonomers bearing a terminal furan moiety and a central triene motif, i.e. bisdienes suitable for DA polycondensations.
This study is an additional contribution to an ongoing research effort aimed at finding novel sources and strategies to valorise plant oils as precursors to original macromolecular materials, particularly with the conjunct incorporation of furan moieties, i.e. another family of structures derived from renewable resources.4 The use of the DA reaction to achieve this goal, a click chemistry interaction particularly suited to furan derivatives as dienes, has opened a promising alternative approach for the exploitation of sustainable natural sources at the service of polymer science and technology.
Scheme 1 sets the stage of this investigation by providing a succinct overview of the mechanisms associated with the two adopted strategies, as explained above.
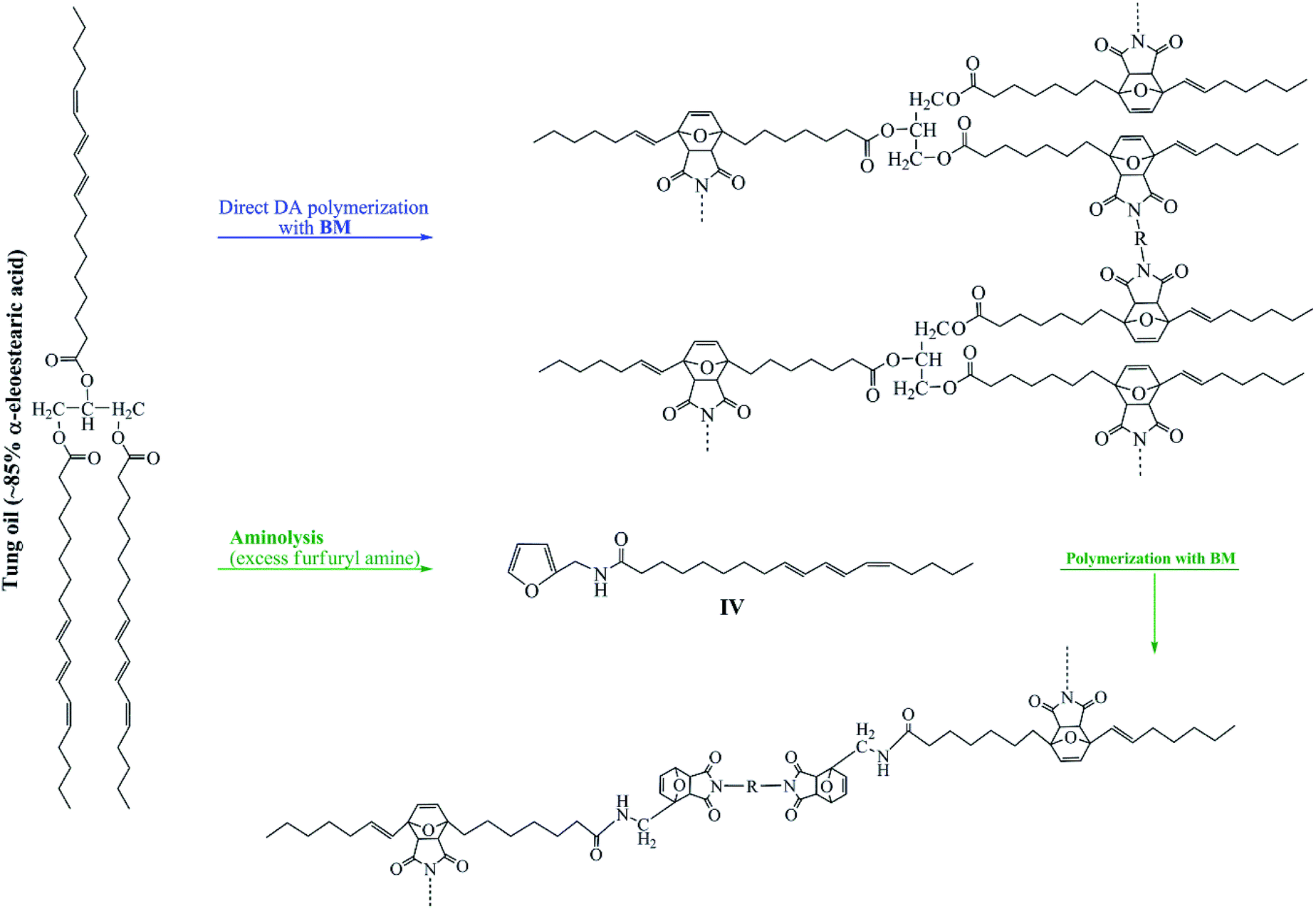 | ||
| Scheme 1 Schematic representation of the use of TO to produce Diels–Alder-based polymers. BM = bismaleimides (Scheme 4). | ||
Experimental section
Materials
Crude TO was purchased from General Iron Fittings Ltda. (São Paulo, Brazil). 1,1′-(Methylenedi-4,1-phenylene)bismaleimide (I), furfuryl amine, maleimide, N-methylmaleimide, 4-toluenesulfonic acid, formaldehyde solution (36.5–38% in H2O), NaOH, furan, maleic anhydride, triethylamine, 1,1,2,2-tetrachloroethane (TCE) and its deuterated counterpart (TCE-d2) were best-purity samples purchased from Sigma-Aldrich company. All solvents and non-solvents were high purity commercial products, which were used as received.Synthesis of N,N′-hexamethylenebismaleimide
The synthesis of N,N′-hexamethylenebismaleimide (II, Scheme 2) was previously reported5 using the classical two-step process involving the reaction of the corresponding diamine with maleic anhydride, followed by the cyclization of the ensuing bismaleamic acid. Here, we adopted the more recent synthetic approach to maleimides based on the reaction of the starting amine with the furan–maleic anhydride DA adduct, followed by the deprotection of the ensuing product to generate the required maleimide (Scheme 2). Hexamethylenediamine (5.8 g, 50.0 mmol) was added to a solution of exo-3,6-epoxy-1,2,3,6-tetrahydrophthalic anhydride (furan–maleic anhydride Diels–Alder adduct) (16.6 g, 100.0 mmol) in methanol (307 mL) with some drops of triethylamine. The solution was stirred at 55–60 °C for 3 days as a precipitate formed progressively. The suspension was filtered and the solid residue washed with cold methanol.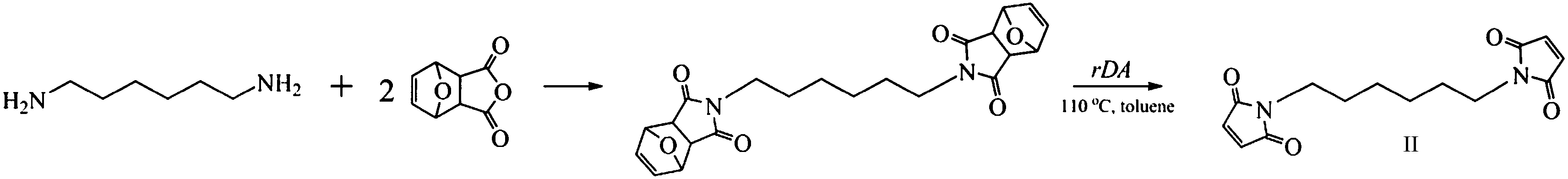 | ||
| Scheme 2 Synthesis of bismaleimide II. | ||
The retro-DA, applied to the final product with the purpose of deprotecting the maleimide groups, was conducted in refluxing toluene for 24 h. This alternative synthetic procedure is more straightforward than that involving the rather delicate cyclization process of the maleamic acid. The structural characterization of the recovered product gave the same spectral features as previously reported.5
Synthesis of bismaleimidomethyl ether
Bismaleimidomethyl ether (III) was prepared according to the procedure described by Tawney and co-workers.16 This synthesis (Scheme 3) called upon the preliminary preparation of hydroxymethylmaleimide by the reaction of maleimide with a formaldehyde basic aqueous solution.16 The spectroscopic characterization of this intermediate was first carried out in the present study, since the original publication only gave its melting temperature (104–106 °C)16 which was the same in our work. The following FTIR and 1H NMR data clearly confirmed the expected structure. 1H NMR (400 MHz, CDCl3, ppm): 6.80 (s, 2H, maleimide protons), 5.11 (d, J = 8 Hz, 2H, methylene protons), 3.06 (t, J = 8 Hz, 1H, N–H proton). FTIR (cm−1): 3404, 3095, 2967, 1698, 1449, 1398, 1364, 1312, 1166, 1029, 918, 841, 755, 696. Mass spectrum [M + Na] = 149 corresponding to C5H4O3NNa, i.e. the expected molecular mass with the loss of the primary alcohol hydrogen atom. | ||
| Scheme 3 Synthesis of bismaleimide III. | ||
The second step consisted in the self-condensation of the ensuing primary alcohol catalyzed by 4-toluenesulfonic acid16 (Scheme 3). Again the obtained bismaleimide III was thoroughly characterized spectroscopically, given that its only available properties were its melting temperature (130–131 °C),16 confirmed in this work with a slightly higher value, and elemental analysis. The spectral data gathered here for the first time, again corroborated the validity of the envisaged structure. 1H NMR (400 MHz, CDCl3, ppm): 6.81 (s, 4H, maleimide protons), 5.08 (s, 4H, methylene protons). FTIR (cm−1): 3095, 2941, 1707, 1584, 1444, 1418, 1342, 1229, 1191, 1097, 1055, 908, 833, 765, 693. Mass spectrum [M + Na] = 259 corresponding to C10H8O5N2Na, i.e. bismaleimide III associated with a sodium atom.
 | ||
| Scheme 4 Structures of the three different bismaleimides used here as dienophiles. | ||
Tung oil reaction with bismaleimides
The reactions of pristine TO with the three different bismaleimides shown in Scheme 4 were carried out separately both in bulk and in TCE at 80 °C under magnetic stirring and a nitrogen atmosphere. The course of these polycondensations was followed by the progressive increase in the medium viscosity and, in the case of crosslinking systems, by the appearance and accumulation of gelled material. The reactions in bulk were limited by the rather low solubility of the bismaleimides in TO. However, three different TO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) : bismaleimide molar ratios were assayed, viz. 1:0.5, 1:1 and 1:1.5, the last leading to the higher crosslinking density, considering the three functionalities present at the TO molecule and the two functionalities of the bismaleimides. The flasks were kept under stirring for three days, and the formation of branched or crosslinked materials was observed in all cases. For the reaction using TCE as solvent, only the TO: bismaleimide ratio of 1:1.5 was assayed. For these reactions, 1.0 mmol TO, 1.5 mmol bismaleimide and 10 mL TCE were added in a round bottom flask, with nitrogen atmosphere. The complete gel formation determined the end of the reaction, which happened after 3 h. The gel was washed thoroughly by magnetic stirring with petroleum ether to eliminate all residual traces of TCE. The product, isolated as a yellowish material, was kept in a vacuum oven at 35 °C until constant weight, leading to a yield of 84%.
: bismaleimide molar ratios were assayed, viz. 1:0.5, 1:1 and 1:1.5, the last leading to the higher crosslinking density, considering the three functionalities present at the TO molecule and the two functionalities of the bismaleimides. The flasks were kept under stirring for three days, and the formation of branched or crosslinked materials was observed in all cases. For the reaction using TCE as solvent, only the TO: bismaleimide ratio of 1:1.5 was assayed. For these reactions, 1.0 mmol TO, 1.5 mmol bismaleimide and 10 mL TCE were added in a round bottom flask, with nitrogen atmosphere. The complete gel formation determined the end of the reaction, which happened after 3 h. The gel was washed thoroughly by magnetic stirring with petroleum ether to eliminate all residual traces of TCE. The product, isolated as a yellowish material, was kept in a vacuum oven at 35 °C until constant weight, leading to a yield of 84%.
Aminolysis of tung oil and its ensuing polymerization with bismaleimides
The bulk reaction of TO with a large excess of furfuryl amine (molar ratio 1:20),2 was conducted at 95 °C for 48 h and led to three identical macromonomers incorporating two different DA-susceptible sites, viz. a furan moiety and the triene from the ensuing α-eleostearic fatty acid amide (IV, Scheme 1). This product (>95% purity) was isolated simply by high vacuum removal of the excess of furfuryl amine, with a yield of 72%. The macromonomers were then polymerized with the three different bismaleimides shown in Scheme 4 under stoichiometric conditions by dissolving the reagents in TCE and keeping the solution under stirring at 70 °C in a nitrogen atmosphere for three days, the reactions being followed by FTIR and 1H NMR spectroscopy. Thereafter, the polymers were precipitated into an excess of cold petroleum ether (40–60 °C fraction), filtered off and dried to constant weight. The yield of the three polymers varied between 65 and 75%.
Macromonomer and polymer characterization
The furan amide macromonomer (IV) and all the polymers were characterized by FTIR and 1H NMR spectroscopy and their thermal properties assessed by TGA with a Perkin Elmer Pyris 1 equipment, working with a nitrogen flow of 20 mL min−1 and a heating rate of 10 °C min−1. The temperature ranged from 25 to 700 °C. DSC analyses were conducted with a Perkin Elmer DSC 8000 equipment, in a nitrogen flow of 20 mL min−1, a heating rate of 10 °C min−1 and a cooling rate of 10 °C min−1. The molecular weight distribution of all soluble polymers was obtained by GPC, using THF as eluent and polystyrene standards. The gels obtained from the direct reaction of TO and bismaleimides were obviously not characterized by 1H NMR.Results and discussion
Tung oil characterization
TO was chosen in this study because of its three conjugated double bonds present in each fatty acid chain, which can act as dienes in Diels–Alder reactions, with maleimides as dienophiles. The choice of a pristine sample was dictated by a working philosophy based on a green chemistry approach. Its spectroscopic characterization revealed the expected structural features, including peaks (IR, cm−1) at ∼3010 for the conjugated double bonds, 2823–2977 for the methylene sequences and ∼1740 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) plus ∼1235 (C–O) for the ester moieties. Its 1H NMR spectrum and the corresponding resonances are shown in Fig. 1, with resonances (ppm) for: (i) one CH3 signal at 0.91, attributed to normal end-of-chain methyl groups; (iia) CH2 sequences centered at 1.33, (iib) the CH2 at the γ-position with respect of the ester carbonyl groups at 1.62, (iic) the CH2 next to a CC unsaturation, centered at 2.13, (iid) the CH2 attached to the ester groups at 2.31, (iie) and those belonging to the glycerol residues at 4.30–4.14 (with the corresponding CH at 5.26); (iii) the protons of the three conjugated double bonds at 5.33–6.45, whose integration in relation to that of the methyl protons gave an average number of double bonds of 8.1 per triglyceride molecule. Given the fact that this was a pristine TO sample, the inevitable presence of small amounts of other triglycerides resulted in relative integration ratios which were modestly different from those expected for the single structure shown in Fig. 1. A previously reported spectrum of TO,14 gave a somewhat lower degree of unsaturation, confirming that the commercial source of the oil can affect its actual composition, which must therefore not be considered as a strictly reproducible feature and justifies the new quantitative examination of the spectrum related to the TO used here.
O) plus ∼1235 (C–O) for the ester moieties. Its 1H NMR spectrum and the corresponding resonances are shown in Fig. 1, with resonances (ppm) for: (i) one CH3 signal at 0.91, attributed to normal end-of-chain methyl groups; (iia) CH2 sequences centered at 1.33, (iib) the CH2 at the γ-position with respect of the ester carbonyl groups at 1.62, (iic) the CH2 next to a CC unsaturation, centered at 2.13, (iid) the CH2 attached to the ester groups at 2.31, (iie) and those belonging to the glycerol residues at 4.30–4.14 (with the corresponding CH at 5.26); (iii) the protons of the three conjugated double bonds at 5.33–6.45, whose integration in relation to that of the methyl protons gave an average number of double bonds of 8.1 per triglyceride molecule. Given the fact that this was a pristine TO sample, the inevitable presence of small amounts of other triglycerides resulted in relative integration ratios which were modestly different from those expected for the single structure shown in Fig. 1. A previously reported spectrum of TO,14 gave a somewhat lower degree of unsaturation, confirming that the commercial source of the oil can affect its actual composition, which must therefore not be considered as a strictly reproducible feature and justifies the new quantitative examination of the spectrum related to the TO used here.
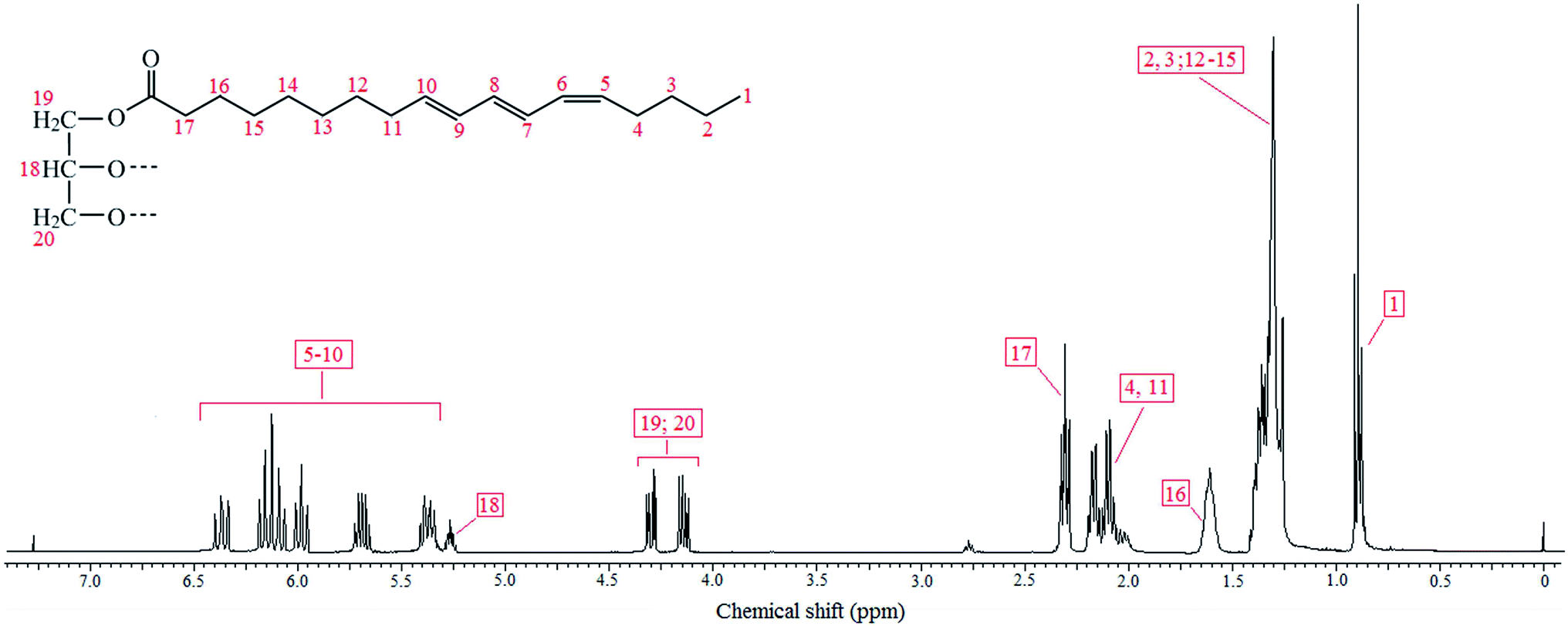 | ||
| Fig. 1 1H NMR spectrum of commercial TO. | ||
Tung oil reactions with maleimides
In order to acquire a clear notion of the DA reaction between the three conjugated double bonds in TO (diene) and a maleimide moiety (dienophile), we first carried out a model reaction involving TO and a monomaleimide, namely, N-methylmaleimide, as previously done with N-phenylmaleimide,14 but under milder conditions. Metzger's group17 studied the reaction of the methyl ester of α-eleostearic acid (the structure encountered in TO) with maleic anhydride in bulk reactions carried out at 150 °C for two hours.This investigation clearly showed the aptitude of the characteristic conjugated structures associated with TO to undergo the DA reaction. In the present context, however, we chose to work with the TO itself and with different dienophiles, which justified this preliminary approach with a monomaleimide.
The reaction was conducted at 65 °C in an NMR tube as a concentrated solution in deuterated tetrachloroethane (TCE-d2), under stoichiometric conditions, viz. 3 moles of N-methylmaleimide per mole of TO. Its progress was followed by 1H NMR spectroscopy and the spectrum taken after three days is shown in Fig. 2, which provides a direct indication of the conversion through the relative intensity of the methyl proton signal relative to the unreacted (2.96 ppm) and reacted (2.81 ppm) N-methylmaleimide, namely about 85% yield. As for the most relevant changes associated with the formation of the DA adduct, they reflect the disappearance of the signals corresponding to the triene moiety at the lower fields above 6 ppm and the corresponding appearance of the ensuing protons on saturated carbons around 3 ppm, apart from the signals between 5.25 and 5.95, reflecting the presence of the unconjugated olefinic protons formed in the reaction (Fig. 2).
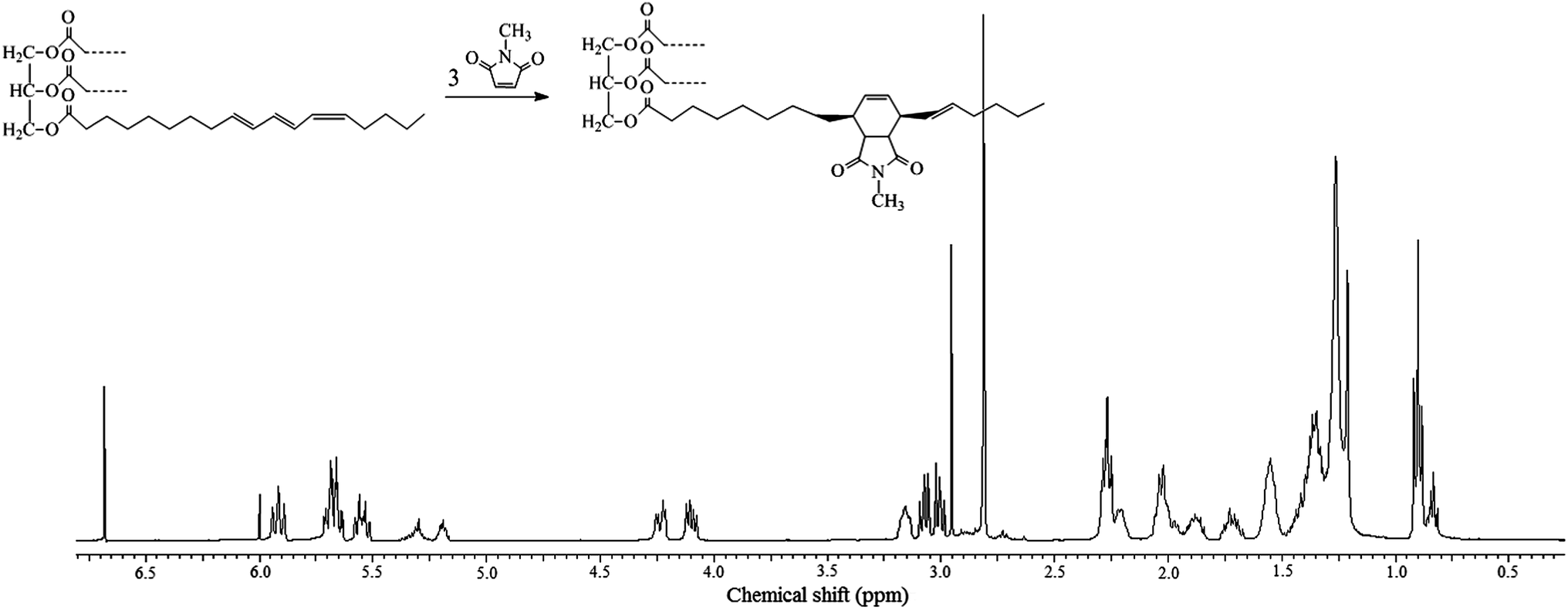 | ||
| Fig. 2 1H NMR spectrum of the reaction product between TO and N-methylmaleimide, showing only one branch of the triglyceride. | ||
This model experiment confirmed the proneness of the three conjugated unsaturations borne by the fatty acid residues of TO to behave as a diene in the DA coupling with malemides. The temperature used here was much lower than those applied previously,14,17 which resulted in a fairly slow reaction, but avoided possible unwanted side reactions. In the subsequent work on polymerizations, we moved therefore to 80 °C in order to accelerate the process, while keeping the temperature well below the onset of free radical mechanisms interfering with the desired DA condensations.
The choice of three different bismaleimides as bisdienophiles applied to the DA polymerizations with TO stems from the idea of preparing macromolecular structures with very different flexibility, the highest rigidity being obviously associated with the aromatic bismaleimide I (Scheme 4).
Most reactions in TCE were conducted with a relative amount of bismaleimide higher than the minimum proportion required to form a network, i.e., [maleimide functions]/[tung oil triene functions] > 0.5, according to Flory–Stockmayer equation applied to a polycondensation system involving a difunctional monomer reacting with a trifunctional counterpart. Whereas bismaleimides I and II were only sparingly soluble in TO, III showed a higher solubility and it was therefore possible to carry out the same reactions in bulk. Although the results were entirely similar, as expected, the interest here is that coating compositions devoid of solvents can be envisaged in which the combined action of DA polycondensation and air siccativity would promote their drying process.
The ensuing gels were thoroughly washed several times with petroleum ether, dried and characterized. Fig. 3 shows typical FTIR spectra of materials arising from each combination, more specifically here when using [bismaleimide]/[TO] = 1, viz. aiming at the highest crosslink density. Whereas the region relative to the methylene sequence is essentially the same for the three networks, the carbonyl region shows the presence of both the TO ester moieties and the maleimide CO. As for the rest of the spectra, the differences arise from the specific structure of each starting bismaleimides with characteristic peaks for 1,4-disubstituted aromatic rings for I, and the C–O–C peaks for III.
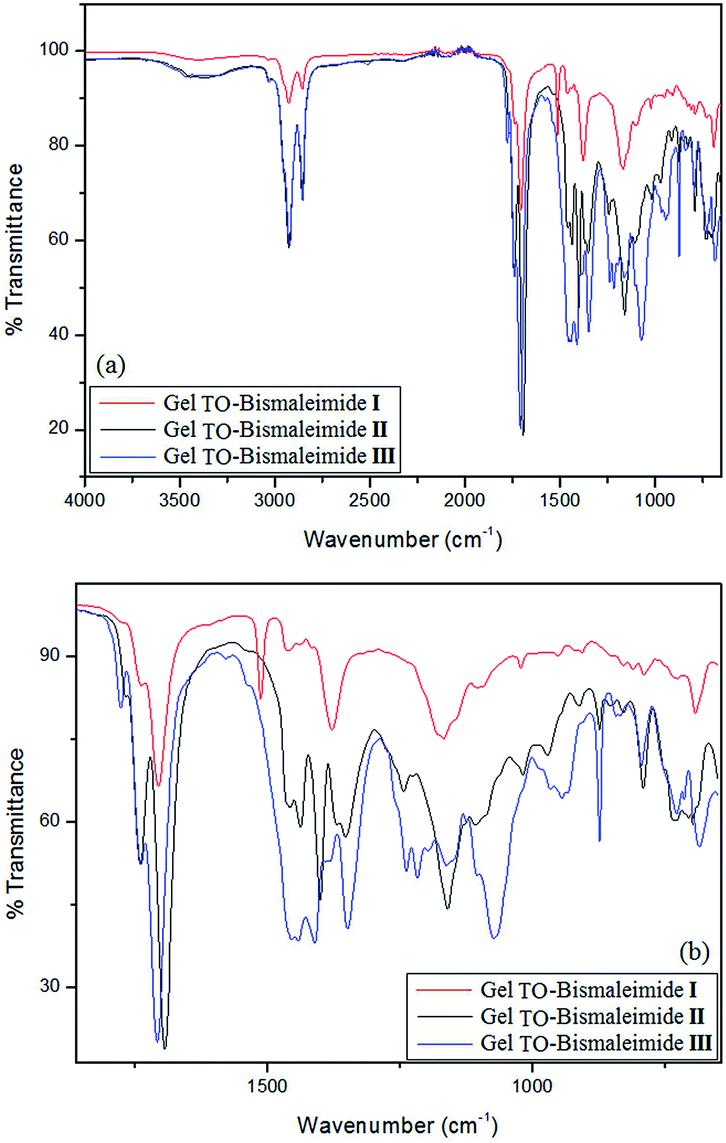 | ||
| Fig. 3 FTIR spectra of typical gels. (a) Full spectra and (b) blowup of the most relevant peak region. | ||
The most relevant point here, however, is the critical fact that the characteristic strong and sharp peak for the in plane maleimide ring deformation, just below 700 cm−1,18 common to all maleimide structures is either absent or very weak, confirming the near complete consumption of each bismaleimide moiety in these non-linear DA polycondensations.
The glass transition temperature of these materials ranged from – 10 °C for TO + II, to – 5 °C for TO + III, to 75 °C for TO + I, in tune with the relative flexibility of the different bridging units in the bismaleimides. As for their thermal stability, the three TGA tracings (Fig. 4) showed the onset of mass loss at about 350 °C, apart from some departure of residual volatiles (solvent/non-solvent trapped inside the gels), with a gradual descent to 550 °C, leaving ∼15% graphitic residue for TO + I, associated with the presence of the aromatic moieties, and less than 3% for the other two gels.
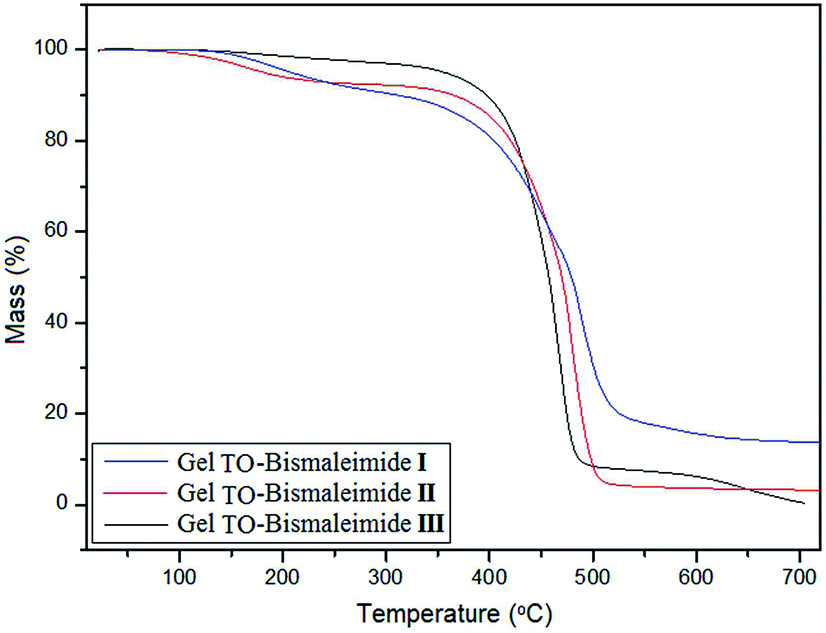 | ||
| Fig. 4 TGA tracings of the three TO-based gels. | ||
The same types of gels were also prepared using bismaleimide III, which was first quickly dissolved in TO at 70 °C, before spreading the ensuing bulk solutions onto various surfaces, including glass, aluminium and Teflon, and placing these liquid films in an oven filled with nitrogen for 24 h at 80 °C. These experiments, which produced completely gelled materials, indicated the viability of performing a surface coating bearing green connotation using exclusively this DA polycondensation. This approach deserves a more thorough investigation.
A number of reactions were also carried out using [maleimide functions]/[tung oil triene functions] molar ratios lower than unity. Given the relative solubility of bismaleimide III in TO, bulk polymerizations with molar ratios of 0.3, 0.5 and 0.75 were conducted at 75 °C for 24 h. Whereas the former only developed a characteristic increase in viscosity, without any precipitate, the other two reactions gave rise to partial gelation, which was much more pronounced with the highest molar ratio. The three ensuing polymers were precipitated in an excess of cold petroleum ether after having removed the gel component, when present. A polycondensation was also carried out in TCE in the same conditions as above, using bismaleimide I, with a molar ratio of 0.3. Again, the system remained homogeneous and the polymer was precipitated and isolated following the same procedure. The precipitated polymers, in all these experiments, displayed a considerable resistance to being redissolved, probably because of their highly branched structure and some tendency to intermolecular association. This unexpected behavior, which precluded making good quality NMR spectra and GPC analyses, is being closely investigated.
Their FTIR spectra were essentially identical to those shown in Fig. 3, in qualitative terms, since neither the degree of branching, nor the average molecular weight would affect the position or the relevant peaks. What varied of course were the relative intensities of peaks arising from the TO and the bismaleimide, as a function of their molar ratio used in each synthesis. The DSC tests on these polymers gave Tg values similar to those given above for the corresponding networks.
It is important to emphasize that within all the reactions described above, TO did not undergo any detectable side reaction, as indeed verified by a blank experiment in which the oil was placed for 48 h at 80 °C in a nitrogen atmosphere without suffering appreciable structural modifications.
While the furan–maleimide DA combination is characterized by a thermal reversibility which takes place typically between 60 and 120 °C, and has therefore been largely exploited in polymer synthesis3 for the preparation of intelligent materials, regrettably this is not the case of the TO–maleimide combination under study. The radical structural change associated with the formation of the adduct in this case is obviously the reason for the lack of the retro-DA reaction, at least within practically accessible temperatures. In other words, the present triene–maleimide DA system possesses the characteristic features of a click chemistry reaction, albeit without the additional advantage of thermal reversibility and therefore the materials described above cannot be deconstructed by heating, as indeed confirmed by their lack of weight loss up to 350 °C, even with the more volatile bismaleimide III.
Tung oil reaction with furfuryl amine and subsequent DA/rDA polymerization
Scheme 1 depicts the strategy followed for this second approach to the exploitation of TO as a source of novel polymeric materials. The completion of the splicing of TO by furfuryl amine, through the aminolysis of its three ester groups, was assessed by the disappearance of the ester carbonyl peak at 1740 cm−1 which had been progressively replaced by the corresponding amide counterpart at 1660 cm−1, as shown in Fig. 5.15 This figure also displays a fairly good isosbestic point close to 1700 cm−1, indicating the occurrence of a single reaction pathway associated with the spectral changes. Concurrently, the 1H NMR signal at 2.30 ppm, attributed to the protons of the methylene group adjacent to the ester moiety in TO (Fig. 1), shifted progressively to 2.20 ppm (Fig. 6), viz. the resonance arising from the protons of the methylene group now attached to the amide moiety.15 | ||
| Fig. 5 The aminolysis of TO by an excess of furfuryl amine as followed by the progressive shift in the infrared carbonyl peak associated with the transformation of the ester moieties into amide counterparts. | ||
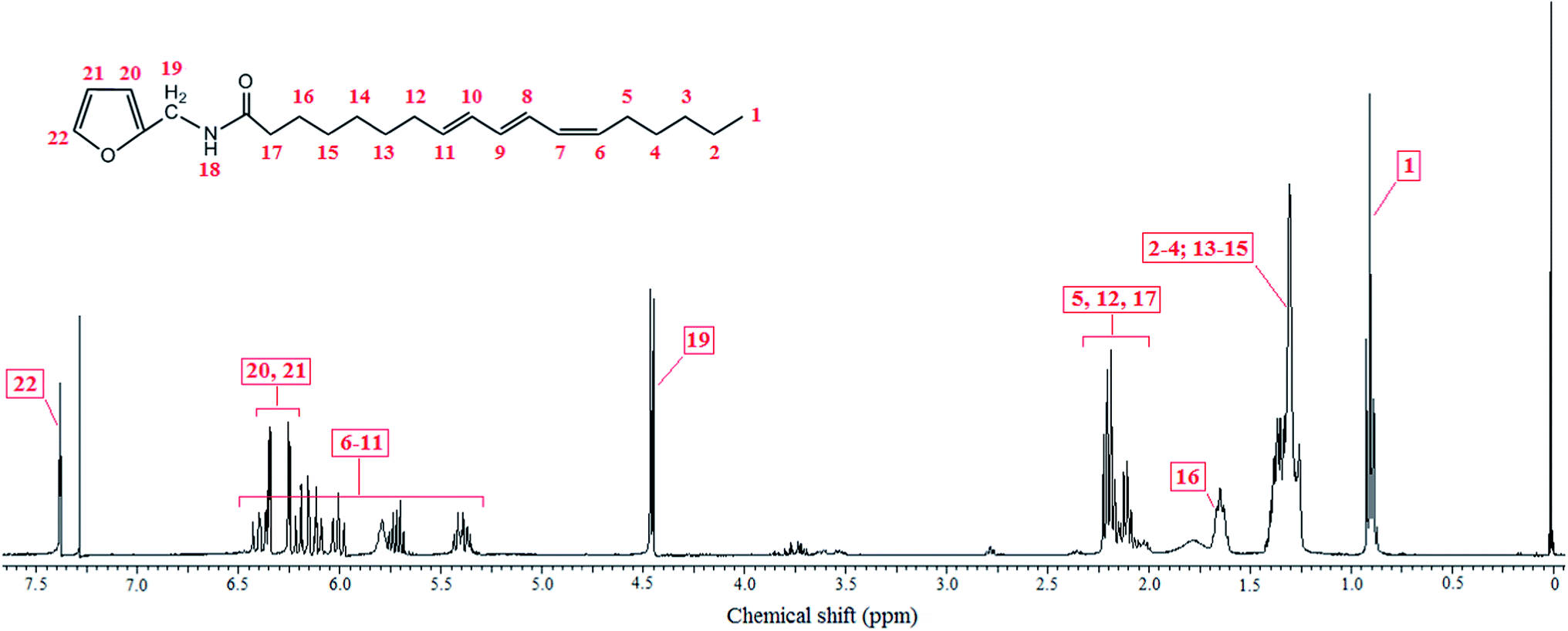 | ||
| Fig. 6 1H NMR spectrum of TO fatty acid furfuryl amide (TOFA, IV). | ||
It is important to recall that this reaction generates a molecule of glycerol for each spliced oil molecule (not shown in Scheme 1), which was subsequently removed, at least in part, by the process of isolation of the main product.
The isolated fatty acid furfuryl amide (TOFA) gave a FTIR spectrum entirely in tune with the expected structure,15 with no evidence of the TO triene moieties having been affected by the reaction. Indeed, the only major changes in the spectrum, compared with that of the starting TO, were the carbonyl shift shown in Fig. 5 and the appearance of the characteristic vibrational modes of the monosubstituted furan heterocycle, particularly the strong ring breathing peak around 1000 cm−1.
The 1H NMR spectrum of TOFA is shown in Fig. 6, together with the assignments, and again, all the other structural features pertaining to the TO chains were clearly preserved. The DSC tracings of TOFA revealed a semi-crystalline character, as confirmed by its birefringence under polarized light optical microscopy, with a melting temperature of 63 °C upon heating and a crystallization temperature of 30 °C upon cooling. Its mass spectrum gave [M + H] = 358, which agrees with TOFA's elemental composition of C23H35O2N (molecular weight of 357).
During the high vacuum removal of the excess of furfuryl amine at temperatures that reached ∼80 °C, most of the glycerol formed in the aminolysis reaction was also removed, but the remainder stayed in the product as displayed in Fig. 6 by the presence of a small multiplet around 3.75 ppm due to the two methylene protons of glycerol.
Given the fact that TOFA is a bifunctional diene DA monomer with one furan and one unsaturated triene moiety, its polycondensations with the three bismaleimides used in this study produced linear macromolecules in which the furan/maleimide adduct was susceptible to undergo the retro-DA reaction, but not the other, as discussed above.
The FTIR spectra of the three DA polycondensates obtained by applying this system are shown in Fig. 7. Apart from the features associated with the specific structure of each bismaleimide, the most relevant point here is the absence of the characteristic furan breathing mode around 1000 cm−1, indicating that near complete yield had been achieved in each reaction. The same conclusion could be drawn from the drastic reduction in the intensity of the peak near 700 cm−1, because of the near absence of maleimide moieties.
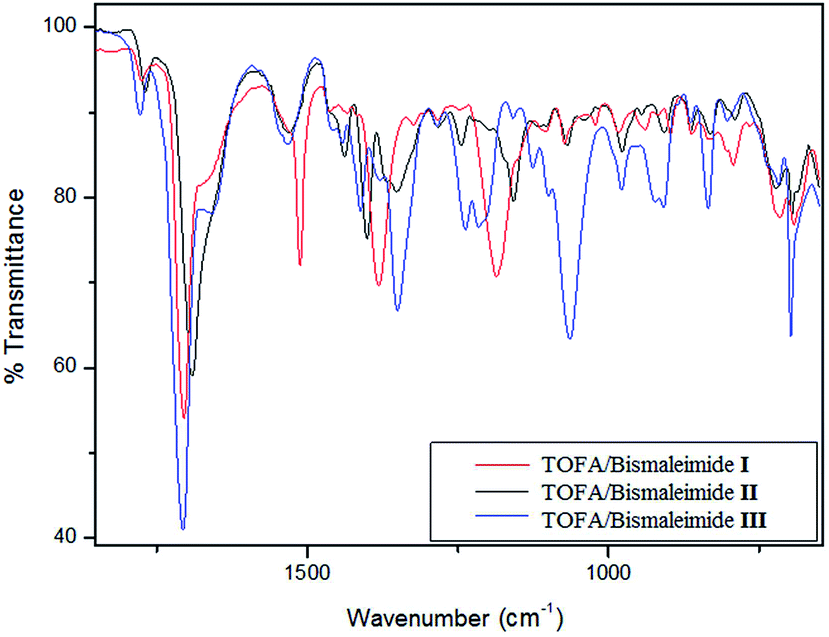 | ||
| Fig. 7 FTIR spectra of the DA polymers prepared from the reaction of TOFA with each bismaleimide. | ||
The 1H NMR spectra of these polymers confirmed the high yield of each polycondensation through the formation of both DA adducts, by the fact that the intensity of the signal at 7.4 ppm, due to the H5 furan proton (Fig. 6), became vanishingly small, and the same applied to the resonance of the maleimide protons at 6.8 ppm, which was replaced by the corresponding furan–maleimide adduct protons at 6.5 ppm. The TGA tracings indicated that their thermal stability reached about 200 °C after which the loss of mass proceeded gradually up to 500 °C, with again a significant carbonaceous residue for the polymer prepared with bismaleimide I. The lower temperature of degradation onset, compared with that of the TO-based gels, was probably due to the occurrence of the retro-DA reaction affecting the furan–maleimide adducts.
The glass transition temperatures of these polycondensates were 105 °C for TOFA + I, to 30 °C for TOFA + II, to 50 °C for TOFA + III, i.e. 40 to 50 °C higher than those of the corresponding TO-based gels. This difference was attributed to the presence of the amide moieties, which are known to establish rather strong interchain hydrogen bonding, compared with the weaker interactions among ester counterparts, as typically encountered between polyamides and polyesters. The GPC of these polymers gave average Mw values ranging between 20000 and 40000 with a DPI always close to two, indicating that all the structures were in fact linear macromolecules.
As pointed out above, these polymers incorporated two types of DA adducts, only one of which, the furan–maleimide structure, was susceptible of undergoing the retro-DA reaction above about 100 °C. The materials were dissolved in TCE-d2 in an NMR tube which was then kept at 120 °C for 24 h, after which spectra were taken and compared with those of the starting polymers (taken in CDCl3). Fig. 8 clearly shows that this thermal treatment gave a high yield of the retro-DA reaction through the reversion of the furan–maleimide adducts, as evidenced by (i) the reappearance of the three furan proton signals around 7.50, 6.25 and 6.40 ppm, (ii) the concomitant near disappearance of the adduct signal at 6.50 ppm, and (iii) the partial reappearance of the maleimide proton signal at 6.75 ppm, keeping in mind that this latter observation only refers to 50% of the reacted maleimide groups, the other 50% being permanently locked into the thermally irreversible TO–maleimide adduct. These features were encountered with all the TOFA polycondensates submitted to this heating treatment, showing that it is possible to depolymerize them, albeit without returning to the corresponding monomers, but rather, statistically, to trimeric oligomers incorporating the TO–maleimide adduct.
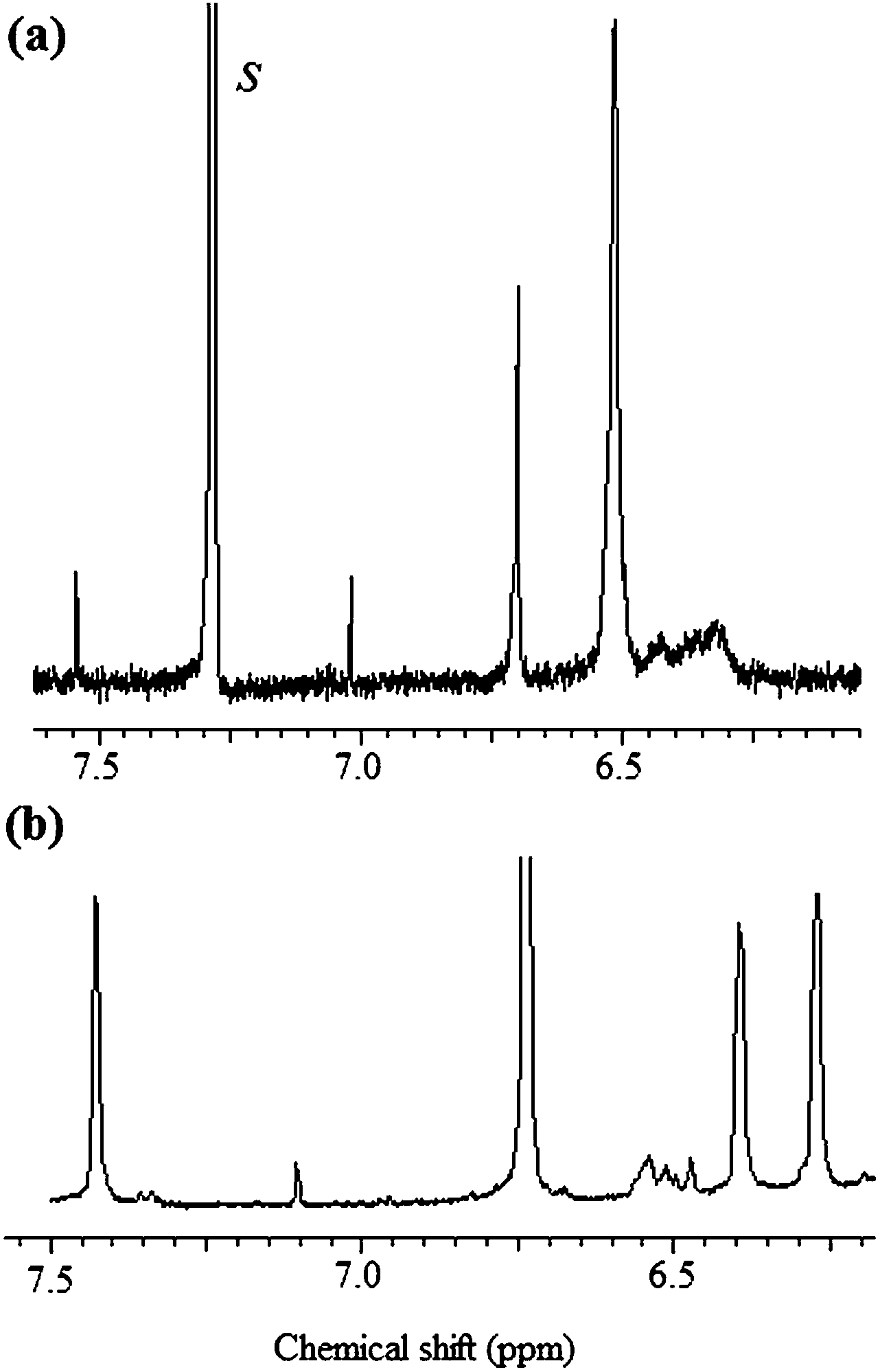 | ||
| Fig. 8 Comparison between the spectrum of polymer TOFA + bismaleimide II before (a) and after (b) the retro-DA reaction. | ||
Conclusions
Tung oil, used as such, and its aminolysis product arising from a straightforward green treatment with furfuryl amine, showed a promising role as Diels–Alder monomers. The properties of the linear, branched and crosslinked materials they generated, derived from the exploitation of renewable resources, could be modulated as a function of the structure of the bismaleimide used as comonomer to give both soft and rigid materials. Their partial depolymerization through the retro-DA reaction could only be applied to the polycondensates prepared from the monomer incorporating furan moieties. The perspective of using these combinations to develop coating formulations without the use of catalysts or solvents represents an additional promising feature provided by this study.Acknowledgements
One of us (TML) wishes to thank FAPESP for her Post-doctoral fellowship (2012/00124-9). AG thanks CAPES for a visiting professorship (707/2012). Thanks also to FAPESP process EMU 2009/54040-8 for MS facilities.Notes and references
- A. Gandini, Green Chem., 2011, 13, 1061 RSC.
- Z. S. Petrović, I. Javni and M. Ionescu, J. Renewable Mater., 2013, 1, 167 CrossRef CAS; R. J. González-Paz, G. Lligadas, J. C. Ronda, M. Galià and V. Cádiz, J. Renewable Mater., 2013, 1, 187 Search PubMed; Y. Xia, R. L. Quirino and R. C. Larock, J. Renewable Mater., 2013, 1, 3 Search PubMed; U. Biermann, U. Bornscheuer, M. A. R. Meier, J. O. Metzger and H. J. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 3854 CrossRef PubMed; O. Türünç and M. A. R. Meier, Macromol. Rapid Commun., 2010, 31, 1822 CrossRef PubMed; M. Galià, L. M. Espinosa, J. C. Ronda, G. Lligadas and V. Cádiz, Eur. J. Lipid Sci. Technol., 2010, 112, 87 CrossRef; Z. S. Petrovic, Polym. Rev., 2008, 48, 109 CrossRef; M. A. R. Meier, J. O. Metzger and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1788 RSC.
- A. Gandini, Prog. Polym. Sci., 2013, 38, 1 CrossRef CAS PubMed.
- C. Vilela, A. J. D. Silvestre and A. Gandini, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2260 CrossRef CAS; C. Vilela, L. Cruciani, A. J. D. Silvestre and A. Gandini, RSC Adv., 2012, 2, 2966 RSC; C. Vilela, L. Cruciani, A. J. D. Silvestre and A. Gandini, Macromol. Rapid Commun., 2011, 32, 1319 CrossRef PubMed.
- A. Gandini, A. Silvestre and D. Coelho, Polym. Chem., 2013, 4, 1364 RSC.
- M. Gomes, A. Gandini, A. J. Silvestre and B. Reis, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3759 CrossRef CAS.
- O. Grosshardt, B. Tübke, K. Kowollik, U. Fehrenbacher, N. Dingenouts and M. Wilhelm, Chem. Ing. Tech., 2009, 81, 1829 CrossRef.
- R.-J. van Putten, J. C. van der Waal, E. Jong, C. B. Rasrendra, H. J. Heeres and J. G. Vries, Chem. Rev., 2013, 113, 1499 CrossRef CAS PubMed.
- C. Liu, X. Yang, J. Cui, Y. Zhou, L. Hu, M. Zhang and H. Liu, BioResources, 2012, 7, 447 CAS.
- C. Meiorin, M. A. Mosiewicki and M. I. Aranguren, Polym. Test., 2013, 32, 249 CrossRef CAS PubMed.
- C. Meiorin, M. I. Aranguren and M. A. Mosiewicki, J. Appl. Polym. Sci., 2012, 124, 5071 Search PubMed; F. Li and R. Larock, J. Appl. Polym. Sci., 2000, 78, 1044 CrossRef CAS.
- Y. Huang, L. Pang, H. Wang, R. Zhong, Z. Zeng and J. Yang, Prog. Org. Coat., 2013, 76, 654 CrossRef CAS PubMed.
- M. A. Mosiewicki, U. Casado, N. E. Marcovich and M. I. Aranguren, Polym. Eng. Sci., 2009, 49, 685 CAS.
- M. Shibata, N. Teramoto and Y. Nakamura, J. Appl. Polym. Sci., 2011, 119, 896 CrossRef CAS.
- A. Gandini, T. M. Lacerda and A. J. F. Carvalho, Green Chem., 2013, 15, 1514 RSC.
- P. O. Tawney, R. H. Snyder, R. P. Conger, K. A. Leibbrand, C. H. Stiteler and A. R. Williams, J. Org. Chem., 1961, 26, 15 CrossRef CAS.
- U. Biermann, W. Butte, T. Eren, D. Haase and J. O. Metzger, Eur. J. Org. Chem., 2007, 23, 3859 CrossRef.
- S. F. Parker, Spectrochim. Acta, Part A, 2006, 63, 544 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2014 |