DOI:
10.1039/C4RA03369H
(Paper)
RSC Adv., 2014,
4, 26524-26534
An unusual Wittig reaction with sugar derivatives: exclusive formation of a 4-deoxy analogue of α-galactosyl ceramide†
Received
14th April 2014
, Accepted 3rd June 2014
First published on 6th June 2014
Abstract
The Wittig reaction of the pyrano-type reducing sugars undergoes an unexpected formation of dienes through the elimination of a benzyloxy group in the presence of t-BuOK. LiHMDS is used rather than t-BuOK to prevent alcohol elimination in the same sugar derivatives. Collectively, t-BuOK has unusual functions in the Wittig reaction that correspond with other bases such as LiHMDS, NaH, and n-BuLi. This unusual function of t-BuOK showed that a unique 4-deoxy-5-hydroxyl analogue 2 of α-galactosyl ceramide was formed exclusively.
Introduction
The Wittig reaction is one of the most commonly used methods for the synthesis of alkenes which has attracted substantial interest from the scientific community.1 The mild reaction conditions, high yields, and the absence of migration for the formed bond are notable features of the Wittig reaction. The remarkable success of the Wittig reaction in synthetic organic chemistry originates from using phosphonium ylides with the formation of phosphonium oxide as a side product, which drives the reaction to completion. The chemistry of the Wittig reaction was initially applied to sugars by Kuhn and Brossmer in 1962, who reacted glyceraldehyde with (carbethoxymethylene) triphenyl-phosphorane.2 Thereafter, notable applications of this reaction were expanded in various directions for approximately 6 decades.3 Usually the carbonyl olefin product is often achieved in normal condition of Wittig reaction. Interestingly, previous reports have demonstrated that 2-deoxy sugars undergoes base promoted β-elimination to form corresponding dienes.4 It is worth mentioning here that there are few reports appeared recently describing the elimination reaction of pyrano-sugar derivatives5 as well as α,β-elimination of anomeric form of pyranoses which provided the dienes under the influence of base6 during the Wittig reaction.
The α-galactosyl ceramide 1 (ref. 7) (α-GalCer) is also referred to as KRN7000 (Fig. 1), and is a commonly known significant compound that is best characterized as an antigen for CD1d-reactive T cells in mice and humans.8 The structure of the 4-deoxy-5-hydroxyl analog 2 (Fig. 1) of α-GalCer is composed of an α-linked D-galacto-pyranoside with phytosphingosine-like ceramide. The hydroxyl groups of α-galactosyl ceramide (α-GalCer) 1 play significant role through hydrogen bonding with various proteins9 that can be recognize with the help of X-ray crystallographic analysis.20
 |
| | Fig. 1 α-Galactosyl ceramide 1 and its 4-deoxy-5-hydroxyl analogue 2. | |
Kim et al. disclosed the role of the 4-hydroxyl group for CD1d-mediated NKT cell activation.10 Van Calenbergh conducted synthesis and in vitro evaluation of α-GalCer epimers.11 Because of its unique structure, no study has reported a method for the synthesis of analogue 2. However, we herein report the straightforward synthesis of 4-deoxy-5-hydroxyl analogue 2 (Fig. 1) by applying an unusual Wittig reaction with reducing disaccharides.
Results and discussion
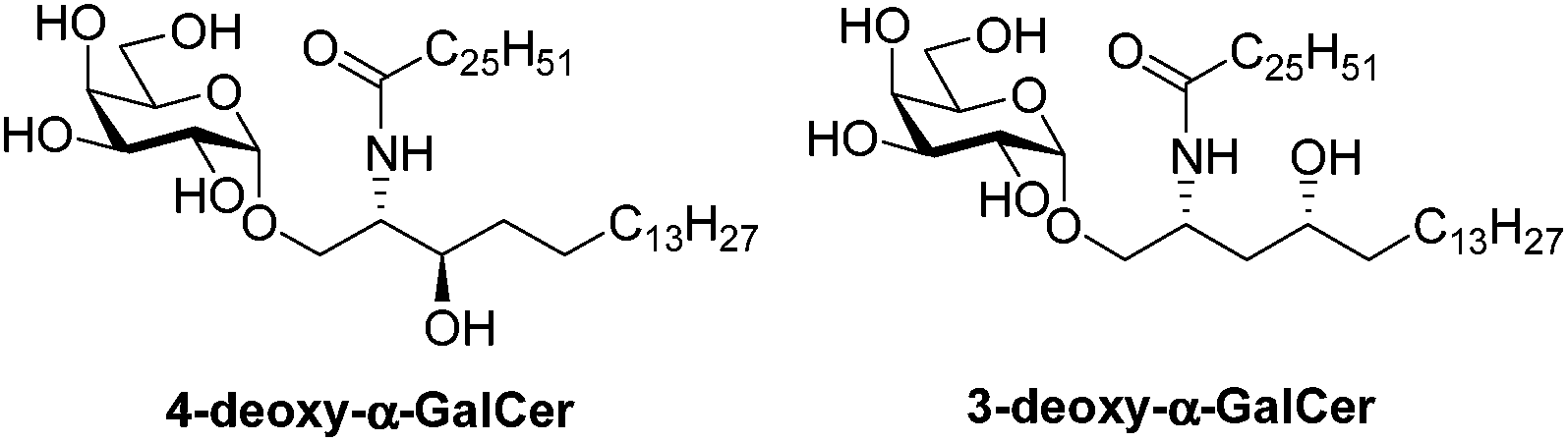
We recently published a concise synthesis of α-galactosyl ceramide from D-galactosyl iodide and D-lyxose12a and conducted a study to prepare the derivatives of α-galactosyl ceramide.12b However, we attempted to prepare α-GalCer analogues in the phytosphingolipid chain that contain one additional hydroxyl group compared to phytosphingosine. Kim and co-workers10 reported the 3-deoxy and 4-deoxy analogues of α-GalCer with results of their biological evaluation but they required additional steps to finalize the synthesis of related analogues by using different precursors (Fig. 2).
 |
| | Fig. 2 α-GalCer analogues reported by Kim et al.10 | |
We began by stereoselectively and regioselectively preparing the key disaccharides (5a–5c) from a well-azeotroped solution of 2,3,4,6-tetra-O-benzyl-D-galactosyl iodide (3) and hexopyranoses (4a–4c).12b The galactosyl iodide 3 (ref. 13) was prepared in situ via the treatment of 2,3,4,6-tetra-O-benzyl-D-galactosyl acetate with iodo-trimethylsilane. The in situ generated 3 was then treated with hexopyranoses (4a–4c) in the presence of N,N-diisopropylethylamine (DIPEA, 1 equiv.) and tetra-n-butylammonium iodide (TBAI, 3 equiv.) in toluene for 1 h at 65 °C, followed by azeotropic distillation, provided α-linked disaccharides (5a–5c) containing protected D-galactose 4a, D-glucose 4b, and D-mannose 4c (ref. 14) (Table 1) in good overall yields.13
Table 1 Effect of various bases on Wittig reaction of disaccharides

|
| Entry |
SM |
Base |
Product (yield) |
| 1 |
5a |
4 equiv. t-BuOK |
6a (96%) |
| 2 |
5a |
3 equiv. t-BuOK |
6a (56%) |
| 3 |
5a |
3 equiv. NaH |
7a (04%) |
| 4 |
5a |
2.5 equiv. n-BuLi |
7a (trace) |
| 5 |
5a |
6 equiv. LiHMDS |
7a (90%) |
| 6 |
5b |
4 equiv. t-BuOK |
6b (66%) |
| 7 |
5b |
6 equiv. LiHMDS |
7b (72%) |
| 8 |
5c |
4 equiv. t-BuOK |
6b (84%) |
| 9 |
5c |
6 equiv. LiHMDS |
7c (72%) |
The Wittig reaction was performed using the in situ generation of phosphorane15 with addition of t-BuOK16 to the well-stirred suspension of D-galactose hemiacetal 5a and phosphonium salt at 0 °C, which produced the undesired product 6a in 3 h in an excellent yield (96%, entry 1, Table 1). The effects of changing bases for the formation of eliminated (6a, 6b) and expected (7a–7c) compounds during the Wittig reaction are shown in Table 1.
We initially expected to obtain the olefination product 7a from the Wittig reaction of the hemiacetal of D-galactose 5a by using t-BuOK, but 1H NMR analysis of the reaction product indicated that one set of benzyl protons disappeared, and one additional double bond proton appeared in the olefin region of the proton spectrum. Moreover, the 13C NMR spectrum showed one additional downfield peak at δ 154 ppm, which belongs to diene C-5 of the sphingosine chain of 6a. The formation of 6a and 6b are probably due to initial base-promoted elimination in the ring-opened isomers of 5a to 5c and furnished the olefination products 6a and 6b. The change in the number of equivalent of t-BuOK from 4 to 3 in the similar reaction resulted in a decreased yield of product to 56% (entry 2, Table 1). Using sodium hydride formed only 4% of the desired product 7a (entry 3, Table 1), and n-butyl lithium resulted in trace amounts of the desired single olefination compound 7a on the TLC plate (entry 4, Table 1). It is worth mentioning here that when we have employed some stronger bases, such as NaH16 and n-BuLi,17 we didn't obtain the unexpected diene derivative. However, the expected olefination products were obtained in very low yields (Table 1).
We then applied the similar Wittig reaction exhibiting the D-galactosyl hemiacetal 5a by using the hindered strong base LiHMDS.18 We obtained the expected olefination compound 7a, in an excellent yield (90%, entry 5, Table 1). This Wittig reaction of hemiacetal 5a with LiHMDS takes 24 hours, because the hemiacetal cannot consume before 24 hours. The Similar results were observed during the Wittig reaction of the hemiacetal of D-glucose 5b and D-mannose 5c when using the base LiHMDS, wherein we formed the expected single olefination compounds 7b and 7c in favorable yields (entry 7 and entry 9, Table 1). Moreover, using t-BuOK in the Wittig reactions of the hemiacetal of D-glucose 5b and D-mannose 5c produced the same unexpected dienes 6b in moderate to favorable yields (entry 6 and entry 8, Table 1). Conversely, this unexpected formation of 4-deoxy sugars occurred through the elimination of the benzyloxy group at the C-3 position during the Wittig reaction of sugar derivatives (5a–5c).
For further study of these unusual Wittig products, we have carried out the Wittig reaction of hexapyranose sugars 8 as shown in Table 2. In this regard, first, we have treated galactosyl hemiacetal 8a with t-BuOK and phosphonium salt in THF at 0 °C (entry 1, Table 2) which provided eliminated compound 9a in 71% isolated yield. Similarly, the reaction of glucosyl hemiacetal 8b (entry 3, Table 2) and mannosyl hemiacetal 8c (entry 5, Table 2) with phosphonium salt and t-BuOK in THF at 0 °C produced eliminated compound 9b in 90 and 79% yield respectively. We have also employed talosyl pyrano-reducing sugar 8d for such kind of transformation under same reaction conditions, which provided eliminated diene 9a and expected olefin 10d in good yields (entry 7 and entry 8, Table 2).
Table 2 Effect of various bases on Wittig reaction of hexopyranoses

|
| Entry |
SM |
Base |
Product (yield) |
| 1 |
8a |
4 equiv. t-BuOK |
9a (71%) |
| 2 |
8a |
6 equiv. LiHMDS |
10a (65%) |
| 3 |
8b |
4 equiv. t-BuOK |
9b (90%) |
| 4 |
8b |
6 equiv. LiHMDS |
10b (72%) |
| 5 |
8c |
4 equiv. t-BuOK |
9b (79%) |
| 6 |
8c |
6 equiv. LiHMDS |
10c (64%) |
| 7 |
8d |
4 equiv. t-BuOK |
9a (80%) |
| 8 |
8d |
6 equiv. LiHMDS |
10d (87%) |
We have also proposed a plausible mechanism for these interesting unexpected transformations as shown in Scheme 1. First, hemiacetal 5a underwent Wittig reaction with phosphonium ylide to provide the olefin 7a via oxa-phosphetane intermediate 11b (pathway a). We assumed that olefin 7a can further undergo β-elimination to provide the diene 6a. In this regard, we have treated olefin 7a with 4 equiv. of t-BuOK and phosphonium salt in THF but we could not obtained any product as the olefin 7a was intact. Therefore, we have proposed alternative scenario where the aldehyde form of 5a can be transformed into α,β-unsaturated aldehyde 11c in presence of t-BuOK, which will be the real reaction partner for the Wittig reaction with phosphonium salt, could provide the unexpected diene 6a according to pathway b.4a
 |
| | Scheme 1 Plausible mechanism of unusual Wittig reaction. | |
We decided to synthesize α-GalCer derivative with diene 6a because of their structural similarities from positions C-2 to C-4 in the α-GalCer 1 and analogue 6a (Scheme 2). We applied Mitsunobu conditions19 to obtain the azido displacement product 12 at the C-2 position, which successfully produced an excellent 96% yield. Because the olefin was reacted with the azido group during intramolecular cycloaddition, we purified azide 12 without data collection and performed the next step directly. The azido compound 12 was used first to form an amine by using the Staudinger reaction followed by an amide bond formation to produce the compound 13 in an 80% yield. Finally, global deprotection obtained an 85% crude yield of the final 4-deoxy-5-hydroxyl analogue 2 by using a palladium catalyst in a chloroform and methanol mixture of solvent at room temperature. At last, the crude product was purified by performing flash column chromatography on silica gel to afford compound 2 in a 62% yield.
 |
| | Scheme 2 Preparation of α-galactosyl ceramide analogue 2. | |
The SAR studies on the crystal structure of α-GalCer complexed with CD1d reveals that the analogue lacking the 4-hydroxyl group on the phytosphingosine exhibits slightly reduced activity as compared to α-GalCer.20 However, the activity of analogue 2 lacking the 4-hydroxyl group, which contains an additional hydroxyl group at the C-5 position of the phytosphingosine chain, remains worthy of investigation. Examining the binding activity of these newly formed compounds with CD1d molecules to stimulate NKT cells would be interesting. Many studies have reported on the biological activities of numerous derivatives with different functional groups at various positions of α-GalCer.20 Based on computer modeling, Henon et al. suggested that the 4-OH group in the sphingosine chain of α-GalCer could form hydrogen bond with Asp80 of CD1d molecule and was important for the recognition of mouse NKT cells.21 However, whether the 4-OH and 5-OH-bonds play a role to anchor the ligand into the binding groove of CD1d molecule is unclear. Moreover, removing the 3-OH and 2-OH groups on the sphingosine chain of α-GalCer caused no response of splenocytes when stimulated with glycolipid-loaded dendritic cells.22 These data suggested that the hydroxyl group on the sphingosine chain of α-GalCer is very important.
To evaluate the biological activities of α-GalCer and analog 2 (Fig. 3), B lymphoma cells overexpressing mouse CD1d/A20-mCD1d was loaded with these glycolipids to stimulate the Va14-expressing mNK1.2 cells to produce IL-2 and the level of IL-2 was determined by ELISA. As shown in Fig. 2, α-GalCer induced the production of IL-2 in a dose-dependent manner (5 μM: 305 ± 7.3 and 25 μM: 404 ± 9.7). IL-2 induction by analog 2 at 5 μM (323.4 ± 27.4) was comparable to α-GalCer, but significantly lower than α-GalCer at 25 μM (322.9 ± 32.3, p < 0.05 by one-way ANOVA with Tukey's multiple comparison test).
 |
| | Fig. 3 Biological activity of α-GalCer analogue 2. The indicated glycolipids (5 μM and 25 μM) were loaded onto A20-mCD1d cells and co-cultured with mNK1.2 cells. Three days after incubation, supernatants were harvested to determine the production of IL-2 by ELISA. Data were presented as means ± SD. * p < 0.05 vs. 25 μM of α-GalCer by one-way ANOVA with Tukey's multiple comparison test. | |
These results suggested that the hydroxyl group at the 4th position of the sphingosine chain of α-GalCer is important for the CD1d α-GalCer complex to stimulate the NKT cells. Whether such modification at the sphingosine chain of α-GalCer could influence the Th1/Th2 polarization awaits future investigation.
Conclusion
In conclusion, the Wittig reaction can be applied to extend the sphingosine chain of α-GalCer. The unexpected formation of a diene analogue enables preparing unique type of α-GalCer 2 derivative. Using the base LiHMDS rather than t-BuOK prevents the formation of unexpected olefination compounds. Otherwise, elimination occurs, and diene is the only product in various pyrano-type reducing sugar derivatives. The mechanism in which t-BuOK influences the outcome of the reaction has been proposed. This unusual effect of t-BuOK has been extended to the hexopyranoses of various sugar derivatives. The hydroxyl group at 4th position of the sphingosine chain in α-GalCer plays an important role for the interaction between CD1d–glycolipid complex and T-cell receptor of NKT cells.
Experimental section
General information
Some reactions were conducted in flame-dried glassware, under nitrogen atmosphere. Dichloromethane, tetrahydrofuran, toluene and N,N-dimethylformamide were purified and dried from a safe purification system containing activated Al2O3. All reagents obtained from commercial sources were used without purification, unless otherwise mentioned. Flash column chromatography was carried out on Silica Gel 60 (230–400 mesh, E. Merck). TLC was performed on pre-coated glass plates of Silica Gel 60 F254 (0.25 mm, E. Merck); detection was executed by spraying with a solution of Ce(NH4)2(NO3)6 (0.5 g), (NH4)6Mo7O24 (24 g) and H2SO4 (28 mL) in water (500 mL) and subsequent heating on a hot plate. Optical rotations were measured at 589 nm (Na) at ∼27 °C. 1H, 13C NMR, DEPT, 1H–1H COSY, 1H–13C COSY, and 1H–1H NOESY spectra were recorded with 400 and 600 MHz instruments. Chemical shifts are in ppm from Me4Si, generated from the CDCl3 lock signal at δ 7.24 ppm. IR spectra were taken with a FT-IR spectrometer using KBr plates. Mass spectra were analyzed on a Finnigan LTQ-OrbitrapxL instrument with an ESI source.
2,3,4-Tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzyl-α-D-galactopyra-nosyl)-D-galactopyranose (5a). To a solution of 1-O-acetyl-2,3,4,6-tetra-O-benzyl-β-D-galactopyranoside 3 (760 mg, 1.31 mmol) in anhydrous dichloromethane (8 mL) was added iodotrimethylsilane (232 μL, 1.63 mmol) at 0 °C under nitrogen atmosphere. After the reaction was stirred for 30 min, the mixture was evaporated in vacuo. Toluene (7 mL) was added to the residue and evaporated in vacuo for three times. In another round bottom flask, a mixture of acceptor 4a (600 mg, 1.33 mmol), diisopropylethylamine (227 μL, 1.31 mmol), tetrabutylammonium iodide (1.44 g, 3.92 mmol) and 4 Å molecular sieves in anhydrous toluene (7 mL) was stirred for 10 min at 65 °C under nitrogen atmosphere. A solution of iodo-residue in toluene (7 mL) was transferred into the reaction flask which contains acceptor, the mixture was kept stirring for 1 h, and ethyl acetate (20 mL) was added to the reaction flask to remove white precipitate and molecular sieves by filtration through celite. The resulting mixture was extracted with aqueous Na2S2O3 (3 × 15 mL) and brine (15 mL), and the organic layers were dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography on silica gel to afford the desired product 5a (960 mg, 75%). Rf 0.5 (EtOAc/Hex = 1/2); [α]29D +38.9 (c 1.2, CHCl3); IR (CHCl3) ν 3445, 3030, 1454, 1098 cm−1; α-form: 1H NMR (400 MHz, CDCl3) δ 7.39–7.23 (m, 35H, ArH), 5.18 (d, J = 2.4 Hz, 1H, H-1), 4.91 (d, J = 10.4 Hz, 1H, CH2Ph), 4.90 (d, J = 11.6 Hz, 1H, CH2Ph), 4.89 (d, J = 10.4 Hz, 1H, CH2Ph), 4.88 (d, J = 12.0 Hz, 1H, CH2Ph), 4.78 (d, J = 11.6 Hz, 2H, CH2Ph), 4.75 (d, J = 3.2 Hz, 1H, H-1′), 4.60 (d, J = 10.4 Hz, 2H, CH2Ph), 4.53 (d, J = 11.6 Hz, 2H, CH2Ph), 4.46 (d, J = 11.6 Hz, 1H, CH2Ph), 4.42 (d, J = 12.0 Hz, 1H, CH2Ph), 4.35 (d, J = 11.6 Hz, 1H, CH2Ph), 4.30 (d, J = 11.6 Hz, 1H, CH2Ph), 4.13 (m, 1H, H-3′), 4.03–3.89 (m, 7H, H-2, H-2′, H-3, H-4, H-4′, H-5, H-5′), 3.62–3.48 (m, 4H, H-6a, H-6b, H-6a′, H-6b′), 3.41 (s, 1H, OH); 13C NMR (150 MHz, CDCl3) δ 138.62 (C), 138.58 (C), 138.5 (C), 138.4 (C × 2), 138.2 (C), 137.5 (C), 128.4 (CH × 2), 128.3 (CH × 4), 128.23 (CH × 2), 128.19 (CH × 2), 128.15 (CH × 4), 128.1 (CH × 2), 128.04 (CH × 2), 128.01 (CH × 2), 127.96 (CH × 2), 127.9 (CH × 2), 127.8 (CH), 127.61 (CH), 127.57 (CH), 127.5 (CH × 2), 127.4 (CH × 2), 127.34 (CH × 2), 127.30 (CH × 2), 98.4 (CH), 91.5 (CH), 78.9 (CH), 78.6 (CH), 76.4 (CH), 76.3 (CH), 75.2 (CH), 74.6 (CH), 74.4 (CH2), 73.5 (CH2), 73.43 (CH2), 73.39 (CH2), 73.3 (CH2), 73.0 (CH2), 72.9 (CH2), 72.8 (CH2), 69.7 (CH), 69.3 (CH), 68.9 (CH2); β-form: 1H NMR (400 MHz, CDCl3) δ 7.37–7.23 (m, 35H, ArH), 4.82–4.63 (m, 15H, H-1′, CH2Ph), 4.56 (d, J = 8.4 Hz, 1H, H-1), 4.03–3.89 (m, 4H, H-2′, H-3, H-3′, H-4′), 3.82–3.68 (m, 4H, H-2, H-5, H-6a, H-6a′), 3.64–3.48 (m, 4H, H-4, H-5′, H-6b, H-6b′), 3.41 (s, 1H, OH); 13C NMR (150 MHz, CDCl3) δ 138.62 (C), 138.58 (C), 138.5 (C), 138.4 (C × 2), 138.2 (C), 137.5 (C), 128.4 (CH × 2), 128.3 (CH × 4), 128.23 (CH × 2), 128.19 (CH × 2), 128.15 (CH × 4), 128.1 (CH × 2), 128.04 (CH × 2), 128.01 (CH × 2), 127.96 (CH × 2), 127.9 (CH × 2), 127.8 (CH), 127.61 (CH), 127.57 (CH), 127.5 (CH × 2), 127.4 (CH × 2), 127.34 (CH × 2), 127.30 (CH × 2), 98.2 (CH), 97.6 (CH), 81.9 (CH), 80.6 (CH), 79.0 (CH), 76.2 (CH), 74.8 (CH2), 74.3 (CH), 74.3 (CH), 74.0 (CH), 73.5 (CH2), 73.43 (CH2), 73.39 (CH2), 73.3 (CH2), 73.0 (CH2), 72.9 (CH2), 72.8 (CH2), 69.4 (CH), 68.4 (CH2); HRMS (ESI, M + Na+) calcd for C61H64O11Na 995.4341, found 995.4380.
2,3,4-Tri-O-benzyl-6-O-(2,3,4,6-tetra-O-benzyl-α-D-galactopyra-nosyl)-D-glucopyranose (5b). To a solution of 1-O-acetyl-2,3,4,6-tetra-O-benzyl-β-D-galactopyranoside 3 (1.90 g, 3.26 mmol) in anhydrous dichloromethane (20 mL) was added iodotrimethylsilane (578 μL, 4.08 mmol) at 0 °C under nitrogen atmosphere. After the reaction was stirred for 30 min, the mixture was evaporated in vacuo. Toluene (17 mL) was added to the residue and evaporated in vacuo for three times. In another round bottom flask, a mixture of acceptor 4b (1.50 g, 3.33 mmol), diisopropylethylamine (567 μL, 3.26 mmol), tetrabutyl-ammonium iodide (3.60 g, 9.78 mmol) and 4 Å molecular sieves in anhydrous toluene (17 mL) was stirred for 10 min at 65 °C under nitrogen atmosphere. A solution of iodo-residue in toluene (17 mL) was transferred into the reaction flask which contains acceptor, the mixture was kept stirring for 1 h, and ethyl acetate (30 mL) was added to the reaction flask to remove white precipitate and molecular sieves by filtration through celite. The resulting mixture was extracted with aqueous Na2S2O3 (3 × 20 mL) and brine (20 mL), and the organic layers were dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography on silica gel to afford the desired product 5b (2.33 g, 72%). Rf 0.5 (EtOAc/Hex = 1/2); [α]29D +39.0 (c 1.3, CHCl3); IR (CHCl3) ν 3431, 3030, 1454, 1096 cm−1; α-form: 1H NMR (400 MHz, CDCl3) δ 7.37–7.24 (m, 35H, ArH), 5.09 (d, J = 2.8 Hz, 1H, H-1), 4.97 (d, J = 3.2 Hz, 1H, H-1′), 4.95–4.53 (m, 12H, CH2Ph), 4.42 (d, J = 12.0 Hz, 1H, CH2Ph), 4.35 (d, J = 12.4 Hz, 1H, CH2Ph), 4.07–4.00 (m, 2H, H-2′, H-5), 3.97–3.83 (m, 4H, H-3, H-3′, H-4′, H-6a′), 3.75–3.68 (m, 1H, H-6b′), 3.54 (m, 4H, H-4, H-5′, H-6a, H-6b), 3.40 (dd, J = 9.2, 3.6 Hz, 1H, H-2); 13C NMR (150 MHz, CDCl3) δ 138.64 (C), 138.59 (C), 138.5 (C), 138.4 (C), 138.1 (C), 137.9 (C), 137.7 (C), 128.3 (CH × 2), 128.24 (CH × 2), 128.18 (CH × 2), 128.11 (CH × 2), 128.09 (CH × 2), 128.07 (CH × 2), 128.0 (CH), 127.82 (CH × 2), 127.78 (CH × 2), 127.74 (CH × 2), 127.72 (CH × 2), 127.71 (CH × 2), 127.69 (CH × 2), 127.67 (CH × 2), 127.65 (CH × 2), 127.6 (CH), 127.54 (CH), 127.49 (CH), 127.42 (CH), 127.37 (CH), 127.31 (CH), 98.2 (CH), 90.7 (CH), 81.6 (CH), 80.1 (CH), 78.2 (CH), 77.9 (CH), 76.5 (CH), 75.5 (CH2), 74.84 (CH2), 74.83 (CH), 74.58 (CH2), 74.56 (CH2), 73.2 (CH2), 72.8 (CH2), 72.7 (CH2), 70.3 (CH), 69.3 (CH), 68.9 (CH2), 67.3 (CH2); β-form: 1H NMR (400 MHz, CDCl3) δ 7.37–7.24 (m, 35H, ArH), 5.00 (d, J = 3.6 Hz, 1H, H-1′), 4.97–4.53 (m, 13H, CH2Ph, H-1), 4.44 (d, J = 11.2 Hz, 1H, CH2Ph), 4.30 (d, J = 11.6 Hz, 1H, CH2Ph), 4.07–3.39 (m, 11H, H-2′, H-3, H-3′, H-4, H-4′, H-5, H-5′, H-6a, H-6b, H-6a′, H-6b′), 3.27 (dd, J = 8.4, 8.0 Hz, 1H, H-2); 13C NMR (150 MHz, CDCl3) δ 138.64 (C), 138.59 (C), 138.5 (C), 138.4 (C), 138.2 (C), 138.1 (C), 137.5 (C), 128.3 (CH × 2), 128.24 (CH × 2), 128.18 (CH × 2), 128.11 (CH × 2), 128.09 (CH × 2), 128.07 (CH × 2), 128.0 (CH), 127.82 (CH × 2), 127.78 (CH × 2), 127.74 (CH × 2), 127.72 (CH × 2), 127.71 (CH × 2), 127.69 (CH × 2), 127.67 (CH × 2), 127.65 (CH × 2), 127.6 (CH), 127.54 (CH), 127.49 (CH), 127.42 (CH), 127.37 (CH), 127.31 (CH), 98.1 (CH), 97.1 (CH), 84.3 (CH), 83.2 (CH), 78.4 (CH), 77.6 (CH), 75.3 (CH2), 75.2 (CH), 74.8 (CH), 74.7 (CH2), 74.5 (CH), 74.3 (CH2), 73.3 (CH2), 73.0 (CH2), 72.7 (CH2), 71.9 (CH2), 71.6 (CH), 69 (CH2), 67.9 (CH2); HRMS (ESI, M + Na+) calcd for C61H64O11Na 995.4341, found 995.4343.
2,3,4-Tri-O-bezyl-6-O-(2,3,4,6-tetra-O-benzyl-α-D-galactopyran-osyl)-D-mannopyranoside (5c). To a solution of 1-O-acetyl-2,3,4,6-tetra-O-benzyl-β-D-galactopyranoside 3 (200 mg, 0.34 mmol) in anhydrous dichloromethane (2 mL) was added iodotrimethylsilane (62 μL, 0.43 mmol) at 0 °C under nitrogen atmosphere. After the reaction was stirred for 30 min, the mixture was evaporated in vacuo. Toluene (2 mL) was added to the residue and evaporated in vacuo for three times. In another round bottom flask, a mixture of acceptor 4c (170 mg, 0.38 mmol), diisopropylethylamine (60 μL, 0.34 mmol), tetrabutylammonium iodide (380 mg, 1.02 mmol) and 4 Å molecular sieves in anhydrous toluene (2 mL) was stirred for 10 min at 65 °C under nitrogen atmosphere. A solution of iodo-residue in toluene (2 mL) was transferred into the reaction flask which contains acceptor, the mixture was kept stirring for 1 h, and ethyl acetate (10 mL) was added to the reaction flask to remove white precipitate and molecular sieves by filtration through celite. The resulting mixture was extracted with aqueous Na2S2O3 (3 × 5 mL) and brine (5 mL), and the organic layers were dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography on silica gel to afford the desired product 5c (229 mg, 69%). Rf 0.5 (EtOAc/Hex = 1/2); [α]29D +36.7 (c 0.9, CHCl3); IR (CHCl3) ν 3422, 3030, 1454, 1097 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.37–7.22 (m, 35H, ArH), 5.13 (s, 1H, H-1), 5.01 (d, J = 3.2 Hz, 1H, H-1′), 4.92 (d, J = 11.6 Hz, 1H, CH2Ph), 4.86 (d, J = 10.8 Hz, 1H, CH2Ph), 4.79 (d, J = 11.6 Hz, 1H, CH2Ph), 4.74–4.59 (m, 7H, CH2Ph), 4.55 (d, J = 10.8 Hz, 1H, CH2Ph), 4.53 (d, J = 11.6 Hz, 1H, CH2Ph), 4.42 (d, J = 11.6 Hz, 1H, CH2Ph), 4.35 (d, J = 12.0 Hz, 1H, CH2Ph), 4.07–4.00 (m, 3H, H-2′, H-4, H-5′), 3.96–3.93 (m, 2H, H-3, H-6a), 3.87 (m, 2H, H-3′, H-4′), 3.78–3.71 (m, 3H, H-2, H-5, H-6b), 3.53 (dd, J = 9.6, 6.4 Hz, 1H, H-6a′), 3.42 (dd, J = 9.6, 6.4 Hz, 1H, H-6b′); 13C NMR (100 MHz, CDCl3) δ 138.8 (C), 138.6 (C), 138.51 (C), 137.46 (C), 138.4 (C), 138.3 (C), 137.6 (C), 128.5 (CH × 2), 128.43 (CH × 2), 128.36 (CH × 5), 128.3 (CH × 4), 128.2 (CH × 2), 128.1 (CH × 2), 128.02 (CH × 2), 128.01 (CH × 2), 127.94 (CH × 2), 127.88 (CH × 2), 127.8 (CH), 127.7 (CH × 2), 127.61 (CH × 2), 127.56 (CH × 2), 127.50 (CH × 2), 127.47 (CH), 98.1 (CH), 92.6 (CH), 79.9 (CH), 78.7 (CH), 76.7 (CH), 75.4 (CH), 75.1 (CH), 75.02 (CH), 74.96 (CH2), 74.7 (CH2), 73.4 (CH2), 73.2 (CH2), 73.0 (CH2), 72.8 (CH2), 72.2 (CH2), 72.0 (CH), 69.4 (CH), 69.3 (CH2), 68.50 (CH2); HRMS (ESI, M + Na+) calcd for C61H64O11Na 995.4341, found 995.4331.
(2S,3S,4Z,6Z)-3,5-di-O-benzyl-1-O-(2,3,4,6-tetra-O-benzyl-α-D-galactopyranosyl)-octadec-4,6-dien-1,2,3,5-tetraol (6a). A mixture of disaccharide 5a (980 mg, 1.00 mmol), tridecanyltriphenylphosphonium bromide (2.06 g, 4.03 mmol) and potassium tert-butoxide (452 mg, 4.03 mmol) in anhydrous THF (10 mL) was stirred at 0 °C under nitrogen. After the reaction mixture was kept stirring for 3 h at 0 °C, water (20 mL) was added to quench the reaction and the mixture was extracted with EtOAc (3 × 30 mL). The combined organic layers were washed with brine, dried over anhydrous MgSO4, filtered, and concentrated under vacuum to give a residue. The residue was purified by column chromatography to give the only diene 6a (979 mg, 96%) as colorless oil. Rf 0.5 (EtOAc/Hex = 1/3); [α]29D +12.9 (c 1.6, CHCl3); IR (CHCl3) ν 3063, 2924, 1652, 1454, 1099 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.36–7.20 (m, 30H, ArH), 5.83 (d, J = 12.0 Hz, 1H, H-6), 5.76–5.70 (m, 1H, H-7), 4.90 (d, J = 11.6 Hz, 1H, CH2Ph), 4.80 (d, J = 3.6 Hz, 1H, H-1′), 4.75 (d, J = 12.0 Hz, 1H, CH2Ph), 4.73–4.66 (m, 5H, CH2Ph, H-4), 4.59 (d, J = 12.0 Hz, 1H, CH2Ph), 4.54 (d, J = 11.6 Hz, 1H, CH2Ph), 4.49–4.37 (m, 4H, CH2Ph, H-3), 4.20 (d, J = 11.6 Hz, 1H, CH2Ph), 4.06 (t, J = 6.4 Hz, 1H, H-5′), 4.01 (dd, J = 9.6, 3.6 Hz, 1H, H2′), 3.90–3.87 (m, 2H, H-3′, H-4′), 3.74–3.69 (m, 1H, H-2), 3.66–3.62 (dd, 1H, H-1a), 3.54 (dd, J = 11.2, 3.2 Hz, 1H, H-1b), 3.50 (d, J = 6.8 Hz, 2H, H-6a′, H-6b′), 3.04 (d, J = 4.4 Hz, 1H, 2-OH), 2.27–2.21 (m, 2H, CH2), 1.40–1.23 (m, 18H, CH2), 0.87 (t, J = 6.8 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 154.0 (C), 138.9 (C), 138.73 (C), 138.68 (C), 138.5 (C), 138.0 (C), 137.6 (CH), 137.4 (C), 128.4 (CH × 2), 128.30 (CH × 2), 128.26 (CH × 2), 128.2 (CH × 2), 128.1 (CH × 2), 128.00 (CH × 2), 127.98 (CH × 2), 127.9 (CH × 2), 127.8 (CH × 2), 127.7 (CH × 2), 127.61 (CH × 2), 127.55 (CH × 2), 127.5 (CH × 2), 127.33 (CH × 2), 127.29 (CH × 2), 123.2 (CH), 110.5 (CH), 98.1 (CH), 79.1 (CH), 76.4 (CH), 75.0 (CH), 74.7 (CH2), 73.8 (CH), 73.4 (CH2), 73.2 (CH2), 73.0 (CH2), 72.9 (CH), 70.9 (CH2), 70.1 (CH2), 70.0 (CH2), 69.2 (CH), 68.8 (CH2), 31.9 (CH2), 29.64 (CH2), 29.63 (CH2), 29.61 (CH2), 29.60 (CH2), 29.5 (CH2), 29.4 (CH2), 29.3 (CH2), 29.2 (CH2), 22.9 (CH2), 14.1 (CH3); HRMS (ESI, M + Na+) calcd for C66H80O9Na 1039.5695, found 1039.5717.
(2S,3R,4Z,6Z)-3,5-Di-O-benzyl-1-O-(2,3,4,6-tetra-O-benzyl-α-D-galactopyranosyl)-octadec-4,6-dien-1,2,3,5-tetraol (6b). A mixture of disaccharide 5c (522 mg, 0.54 mmol), tridecanyltriphenylphosphonium bromide (1.09 g, 2.15 mmol) and potassium tert-butoxide (240 mg, 2.15 mmol) in anhydrous THF (5 mL) at 0 °C under nitrogen was stirred. After the reaction mixture was kept stirring for 3 h at 0 °C, water (10 mL) was added to quench the reaction and the mixture was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with brine, dried over anhydrous MgSO4, filtered, and concentrated under vacuum to give a residue. The residue was purified by column chromatography to give the diene 6b (457 mg, 84%) as colorless oil. Rf 0.5 (EtOAc/Hex = 1/3); [α]24D +35.13 (c 0.8, CHCl3); IR (CHCl3) ν 3063, 2924, 1608, 1454, 1059 cm−1; 1H NMR (600 MHz, CDCl3) δ 7.36–7.20 (m, 30H, ArH), 5.83 (d, J = 12.0 Hz, 1H, H-6), 5.73 (dt, J = 12.0, 7.2 Hz, 1H, H-7), 4.91 (d, J = 11.4 Hz, 1H, CH2Ph), 4.80–4.71 (m, 6H, CH2Ph, H-1′, H-4), 4.67 (d, J = 12.0 Hz, 1H, CH2Ph), 4.65 (d, J = 10.2 Hz, 1H, CH2Ph), 4.55 (d, J = 11.4 Hz, 1H, CH2Ph), 4.46–4.42 (m, 3H, CH2Ph, H-3), 4.37 (d, J = 12.0 Hz, 1H, CH2Ph), 4.21 (d, J = 12.0 Hz, 1H, CH2Ph), 4.06–4.01 (m, 2H, H-2′, H-5′), 3.93–3.85 (m, 3H, H-2, H-3′, H-4′), 3.70–3.62 (m, 2H, H-1a, H-1b), 3.49 (d, J = 6.6 Hz, 2H, H-6a′, H-6b′), 3.04 (bs, 1H, 2-OH), 2.28–2.24 (m, 2H, H-8a, H-8b), 1.41–1.20 (m, 18H, CH2), 0.87 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (150 MHz, CDCl3) δ 154.1 (C), 138.8 (C), 138.7 (C), 138.6 (C), 138.3 (C), 137.9 (C), 137.5 (C × 1, CH × 1), 128.34 (CH × 2), 128.31 (CH × 3), 128.19 (CH × 2), 128.15 (CH × 3), 128.04 (CH × 2), 127.96 (CH × 2), 127.82 (CH × 2), 127.77 (CH × 2), 127.64 (CH × 2), 127.61 (CH × 2), 127.5 (CH × 2), 127.4 (CH × 2), 127.33 (CH × 2), 127.25 (CH × 2), 123.2 (CH), 111.1 (CH), 98.2 (CH), 79.1 (CH), 76.3 (CH), 74.8 (CH), 74.7 (CH2), 73.9 (CH), 73.5 (CH2), 73.3 (CH2), 72.9 (CH2), 72.3 (CH), 71.1 (CH2), 70.3 (CH2), 70.0 (CH2), 69.3 (CH), 68.7 (CH2), 31.9 (CH2), 29.62 (CH2 × 2), 29.59 (CH2 × 2), 29.55 (CH2 × 2), 29.4 (CH2), 29.3 (CH2), 29.2 (CH2), 14.1 (CH3); HRMS (ESI, M + Na+) calcd for C66H80O9Na 1039.5695, found 1039.5732.
(2R,3S,4R,5S)-3,4,5-Tri-O-benzyl-1-O-(2,3,4,6-tetra-O-benzyl-α-D-galactopyranosyl)-octadec-6-en-1,2,3,4,5-pentaol (7a). A mixture of disaccharide 5a (1.00 g, 1.03 mmol) and tridecanyl-triphenylphosphonium bromide (3.15 g, 6.17 mmol) in anhydrous tetrahydrofuran (10 mL) was cooled to 0 °C under nitrogen. A 1.0 M solution of lithium hexamethyldisilylamide in THF (6.2 mL, 6.17 mmol) was added to the reaction mixture and the reaction solution was stirred for 24 h at 0 °C. Water (20 mL) was added to quench the reaction and the mixture was extracted with EtOAc (3 × 30 mL). The combined organic layers were washed with brine, dried over anhydrous MgSO4, filtered, and concentrated under vacuum to give a residue. The residue was purified by column chromatography to give the olefin 7a (1.05 g, 90%, cis–trans = 1.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.6) as colorless oil. Rf 0.3 (EtOAc/Hex = 1/4); [α]28D +46.8 (c 1.6, CHCl3); IR (CHCl3) ν 3483, 2925, 2360, 1497, 1061 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.34–7.15 (m, 70H), 5.72 (dt, J = 15.6, 6.8 Hz, 1H), 5.58 (dt, J = 11.6, 7.2 Hz, 1H), 5.50–5.44 (m, 2H), 4.91 (d, J = 11.2 Hz, 2H), 4.83 (d, J = 4.8 Hz, 2H), 4.18–4.64 (m, 11H), 4.60 (d, J = 12.0 Hz, 3H), 4.54 (d, J = 11.2 Hz, 2H), 4.47–4.44 (m, 3H), 4.41 (d, J = 11.2 Hz, 3H), 4.35 (d, J = 11.6 Hz, 2H), 4.27 (d, J = 11.6 Hz, 3H), 4.14–4.07 (m, 2H), 4.05 (m, 5H), 3.93–3.89 (m, 4H), 3.81–3.71 (m, 6H), 3.57 (dd, J = 10.4, 5.6 Hz, 2H), 3.48 (d, J = 6.4 Hz, 4H), 3.26–3.21 (m, 2H), 2.09–1.95 (m, 4H), 1.38–1.95 (m, 36H), 0.88 (t, J = 6.0 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 138.8 (C × 2), 138.6 (C × 2), 138.51 (C × 2), 138.48 (C × 2), 138.33 (C), 137.32 (C), 138.26 (C), 138.2 (C), 137.92 (C), 137.90 (C), 136.0 (CH), 135.0 (CH), 128.36 (CH × 5), 128.35 (CH × 5), 128.3 (CH × 7), 128.24 (CH × 5), 128.21 (CH × 3), 128.19 (CH × 3), 128.15 (CH × 3), 128.07 (CH × 3), 127.06 (CH × 3), 127.9 (CH × 3), 127.8 (CH × 3), 127.7 (CH × 3), 127.60 (CH × 3), 127.57 (CH × 3), 127.52 (CH × 3), 127.48 (CH × 3), 127.45 (CH × 3), 127.42 (CH × 3), 127.35 (CH × 3), 127.3 (CH × 3), 98.12 (CH), 98.06 (CH), 82.06 (CH), 82.04 (CH), 79.9 (CH), 79.2 (CH), 79.1 (CH), 77.40 (CH), 76.3 (CH), 75.3 (CH2), 75.2 (CH2), 74.9 (CH × 3), 74.78 (CH2), 74.76 (CH2), 73.9 (CH), 73.5 (CH2), 73.44 (CH2 × 2), 73.40 (CH2), 73.34 (CH2), 73.32 (CH2), 73.0 (CH2), 72.9 (CH2), 70.4 (CH2), 70.3 (CH2), 69.80 (CH2), 69.78 (CH2), 69.5 (CH), 69.42 (CH × 2), 69.37 (CH), 68.9 (CH2), 68.8 (CH2), 32.4 (CH2), 32.0 (CH2 × 3), 29.7 (CH2 × 5), 29.59 (CH2), 29.55 (CH2), 29.5 (CH2), 29.4 (CH2), 29.39 (CH2), 29.3 (CH2), 29.2 (CH2), 28.1 (CH2), 22.7 (CH2 × 3), 14.2 (CH3 × 2); HRMS (ESI, M + Na+) calcd for C73H88O10Na 1147.6270, found 1147.6261.
:1.6) as colorless oil. Rf 0.3 (EtOAc/Hex = 1/4); [α]28D +46.8 (c 1.6, CHCl3); IR (CHCl3) ν 3483, 2925, 2360, 1497, 1061 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.34–7.15 (m, 70H), 5.72 (dt, J = 15.6, 6.8 Hz, 1H), 5.58 (dt, J = 11.6, 7.2 Hz, 1H), 5.50–5.44 (m, 2H), 4.91 (d, J = 11.2 Hz, 2H), 4.83 (d, J = 4.8 Hz, 2H), 4.18–4.64 (m, 11H), 4.60 (d, J = 12.0 Hz, 3H), 4.54 (d, J = 11.2 Hz, 2H), 4.47–4.44 (m, 3H), 4.41 (d, J = 11.2 Hz, 3H), 4.35 (d, J = 11.6 Hz, 2H), 4.27 (d, J = 11.6 Hz, 3H), 4.14–4.07 (m, 2H), 4.05 (m, 5H), 3.93–3.89 (m, 4H), 3.81–3.71 (m, 6H), 3.57 (dd, J = 10.4, 5.6 Hz, 2H), 3.48 (d, J = 6.4 Hz, 4H), 3.26–3.21 (m, 2H), 2.09–1.95 (m, 4H), 1.38–1.95 (m, 36H), 0.88 (t, J = 6.0 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 138.8 (C × 2), 138.6 (C × 2), 138.51 (C × 2), 138.48 (C × 2), 138.33 (C), 137.32 (C), 138.26 (C), 138.2 (C), 137.92 (C), 137.90 (C), 136.0 (CH), 135.0 (CH), 128.36 (CH × 5), 128.35 (CH × 5), 128.3 (CH × 7), 128.24 (CH × 5), 128.21 (CH × 3), 128.19 (CH × 3), 128.15 (CH × 3), 128.07 (CH × 3), 127.06 (CH × 3), 127.9 (CH × 3), 127.8 (CH × 3), 127.7 (CH × 3), 127.60 (CH × 3), 127.57 (CH × 3), 127.52 (CH × 3), 127.48 (CH × 3), 127.45 (CH × 3), 127.42 (CH × 3), 127.35 (CH × 3), 127.3 (CH × 3), 98.12 (CH), 98.06 (CH), 82.06 (CH), 82.04 (CH), 79.9 (CH), 79.2 (CH), 79.1 (CH), 77.40 (CH), 76.3 (CH), 75.3 (CH2), 75.2 (CH2), 74.9 (CH × 3), 74.78 (CH2), 74.76 (CH2), 73.9 (CH), 73.5 (CH2), 73.44 (CH2 × 2), 73.40 (CH2), 73.34 (CH2), 73.32 (CH2), 73.0 (CH2), 72.9 (CH2), 70.4 (CH2), 70.3 (CH2), 69.80 (CH2), 69.78 (CH2), 69.5 (CH), 69.42 (CH × 2), 69.37 (CH), 68.9 (CH2), 68.8 (CH2), 32.4 (CH2), 32.0 (CH2 × 3), 29.7 (CH2 × 5), 29.59 (CH2), 29.55 (CH2), 29.5 (CH2), 29.4 (CH2), 29.39 (CH2), 29.3 (CH2), 29.2 (CH2), 28.1 (CH2), 22.7 (CH2 × 3), 14.2 (CH3 × 2); HRMS (ESI, M + Na+) calcd for C73H88O10Na 1147.6270, found 1147.6261.
(2R,3R,4R,5S,6Z)-3,4,5-Tri-O-benzyl-1-O-(2,3,4,6-tetra-O-benzyl-α-D-galactopyranosyl)-octadec-6-en-1,2,3,4,5-pentaol (7b). A mixture of disaccharide 5b (730 mg, 0.75 mmol) and tridecanyltriphenylphosphonium bromide (2.30 g, 4.50 mmol) in anhydrous tetrahydrofuran (7 mL) was cooled to 0 °C under nitrogen. A 1.0 M solution of lithium hexamethyldisilylamide in THF (4.50 mL, 4.50 mmol) was added to the mixture and the reaction solution was stirred for 24 h at 0 °C. Water (10 mL) was added to quench the reaction and the mixture was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with brine, dried over anhydrous MgSO4, filtered, and concentrated under vacuum to give a residue. The residue was purified by column chromatography to give the olefin 7b (610 mg, 72%) as colorless oil. Rf 0.3 (EtOAc/Hex = 1/4); [α]27D +51.2 (c 1.1, CHCl3); IR (CHCl3) ν 3463, 3030, 2925, 1497, 1061 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.38–7.22 (m, 35H, ArH), 5.68–5.58 (m, 1H, H-7), 5.40 (t, J = 10.8 Hz, 1H, H-6), 4.92 (d, J = 11.4 Hz, 1H, CH2Ph), 4.86 (d, J = 3.6 Hz, 1H, H-1′), 4.82–4.51 (m, 11H, CH2Ph, H-5), 4.42 (d, J = 11.6 Hz, 1H, CH2Ph), 4.37 (d, J = 12.0 Hz, 1H, CH2Ph), 4.34 (d, J = 12.0 Hz, 1H, CH2Ph), 4.04 (dd, J = 9.6, 3.6 Hz, 1H, H-2′), 4.01–3.97 (m, 2H, H-2, H-5′), 3.95–3.90 (m, 2H, H-3, H-4), 3.81–3.68 (m, 4H, H-1a, H-1b, H-3′, H-4), 3.55–3.44 (m, 2H, H-6a′, H-6b′), 2.08–1.85 (m, 2H, H-8a, H-8b), 1.31–1.17 (m, 18H, CH2), 0.88 (t, J = 6.8 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 138.7 (C), 138.63 (C), 138.60 (C), 138.57 (C × 2), 138.3 (C), 137.8 (C), 135.8 (CH), 128.4 (CH), 128.3 (CH × 5), 128.21 (CH), 128.19 (CH × 2), 128.16 (CH × 4), 128.1 (CH × 5), 128.00 (CH), 127.98 (CH × 2), 127.9 (CH), 127.79 (CH), 127.76 (CH × 2), 127.68 (CH), 127.66 (CH), 127.53 (CH), 127.49 (CH), 127.44 (CH), 127.38 (CH × 3), 127.33 (CH), 127.30 (CH), 126.7 (CH), 98.7 (CH), 81.8 (CH), 79.01 (CH), 78.84 (CH), 76.4 (CH), 75.5 (CH), 74.9 (CH2), 74.8 (CH), 74.7 (CH2), 73.5 (CH2), 73.4 (CH2), 73.0 (CH2), 72.9 (CH2), 70.7 (CH2), 70.2 (CH2), 70.1 (CH), 70.0 (CH), 68.8 (CH2), 31.9 (CH2), 29.7 (CH2), 29.64 (CH2), 29.63 (CH2), 29.61 (CH2), 29.5 (CH2), 29.43 (CH2), 29.3 (CH2), 28.2 (CH2), 22.7 (CH2), 14.1 (CH3); HRMS (ESI, M + Na+) calcd for C73H88O10Na 1147.6270, found 1147.6296.
(2R,3R,4R,5R,6Z)-3,4,5-Tri-O-benzyl-1-O-(2,3,4,6-tetra-O-benzyl-α-D-galactopyranosyl)-octadec-6-en-1,2,3,4,5-pentaol (7c). A mixture of disaccharide 5c (903 mg, 0.92 mmol) and tridecanyltriphenylphosphonium bromide (2.84 g, 5.57 mmol) in anhydrous tetrahydrofuran (10 mL) was cooled to 0 °C under nitrogen. A 1.0 M solution of lithium hexamethyldisilylamide in THF (5.6 mL, 5.6 mmol) was added to the mixture and the reaction solution was stirred for 24 h at 0 °C. Water (10 mL) was added to quench the reaction and the mixture was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with brine, dried over anhydrous MgSO4, filtered, and concentrated under vacuum to give a residue. The residue was purified by column chromatography to give the olefin 7c (726 mg, 70%) as colorless oil. Rf 0.3 (EtOAc/Hex = 1/4); [α]27D +42.6 (c 1.1, CHCl3); IR (CHCl3) ν 3400, 3089, 2924, 1455, 1095 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.34–7.16 (m, 35H, ArH), 5.81–5.75 (m, 1H, H-7), 5.45 (dd, J = 11.2, 9.6 Hz, 1H, H-6), 4.91 (d, J = 11.6 Hz, 1H, CH2Ph), 4.87 (d, J = 3.6 Hz, 1H, H-1′), 4.82 (d, J = 11.6 Hz, 1H, CH2Ph), 4.73 (d, J = 11.6 Hz, 1H, CH2Ph), 4.67 (d, J = 12.0 Hz, 1H, CH2Ph), 4.66 (d, J = 12.0 Hz, 1H, CH2Ph), 4.63–4.53 (m, 6H, CH2Ph), 4.50 (t, J = 9.2 Hz, 1H, H-5), 4.42 (d, J = 12.0 Hz, 1H, CH2Ph), 4.34 (d, J = 12.0 Hz, 1H, CH2Ph), 4.13 (d, J = 11.6 Hz, 1H, CH2Ph), 4.06–4.00 (m, 4H, H-2′, H-3′, H-4′, H-5′), 3.95–3.82 (m, 2H, H-2, H-3), 3.81–3.75 (m, 3H, H-1a, H-1b, H-4), 3.51–3.44 (m, 2H, H-6a′, H-6b′), 2.20–2.01 (m, 2H, H-8a, H-8b), 1.40–1.25 (m, 18H, CH2), 0.88 (t, J = 6.4 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 138.67 (C), 138.66 (C × 2), 138.54 (C), 138.52 (C), 138.1 (C), 137.8 (C), 136.7 (CH), 128.37 (CH × 3), 128.3 (CH × 3), 128.19 (CH × 6), 128.18 (CH × 4), 128.1 (CH), 128.0 (CH × 4), 127.8 (CH), 127.70 (CH × 3), 127.65 (CH), 127.60 (CH × 3), 127.54 (CH), 127.48 (CH), 127.41 (CH × 2), 127.38 (CH), 127.33 (CH), 127.26 (CH), 99.1 (CH), 80.6 (CH), 79.3 (CH), 78.8 (CH), 76.4 (CH), 74.7 (CH2 × 1, CH × 1), 74.5 (CH2), 73.9 (CH2), 73.7 (CH2), 73.6 (CH), 73.4 (CH2), 72.9 (CH2), 71.4 (CH2), 69.73 (CH), 69.69 (CH), 69.4 (CH2), 68.8 (CH2), 31.9 (CH2), 29.66 (CH2), 29.64 (CH2), 29.63 (CH2), 29.61 (CH2), 29.5 (CH2), 29.4 (CH2), 29.3 (CH2), 28.1 (CH2), 22.7 (CH2), 14.1 (CH3); HRMS (ESI, M + Na+) calcd for C73H88 O10Na1147.6270, found 1147.6305.
(2R,3R,4Z,6Z)-1,3,5-Tri-O-benzyl-octadec-4,6-diene-1,2,3,5 pentaol (9a). Method A: a solution of hemiacetal 8a (100 mg, 0.19 mmol) and dodecyltriphenylphosphonium bromide (378 mg, 0.74 mmol) in anhydrous tetrahydrofuran (1 mL) was cooled to 0 °C, followed by quick addition of potassium tert-butoxide (83 mg, 0.74 mmol). After stirring for 1 h at this temperature, the reaction mixture was treated with water (2 mL) and extracted with ethyl acetate (3 × 2 mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated. The residue was purified by column chromatography to afford olefin 9a (77 mg, 71%) as a colorless oil. Method B: a solution of hemiacetal 8d (110 mg, 0.20 mmol) and dodecyltriphenylphosphonium bromide (416 mg, 0.81 mmol) in anhydrous tetrahydrofuran (1.1 mL) was cooled to 0 °C, followed by quick addition of potassium tert-butoxide (91 mg, 0.81 mmol). After stirring for 1 h at this temperature, the reaction mixture was treated with water (1.2 mL) and extracted with ethyl acetate (3 × 2 mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated. The residue was purified by column chromatography to afford olefin 9a (95 mg, 80%) as a colorless oil. Rf 0.56 (EtOAc/Hex = 1/3); [α]22D −9.1 (c 0.8, CH2Cl2); IR (CH2Cl2) ν 2925, 2854, 1729, 1652, 1497, 1455 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.61–7.24 (m, 15H, ArH), 5.86 (d, J = 11.6 Hz, 1H, H-6), 5.76 (dt, J = 11.6, 7.2 Hz, 1H, H-7), 7.41 (s, 2H, CH2Ph), 4.67 (d, J = 9.2 Hz, 1H, H-4), 4.54 (d, J = 12.0 Hz, 1H, CH2Ph), 4.49 (d, J = 12.0 Hz, 1H, CH2Ph), 4.44 (d, J = 11.6 Hz, 1H, CH2Ph), 4.45–4.41 (m, 1H, H-3), 4.25 (d, J = 11.6 Hz, 1H, CH2Ph), 3.72 (dddd, J = 10.4, 7.2, 3.6, 3.6 Hz, 1H, H-2), 3.50 (dd, J = 10.0, 3.6 Hz, 1H, H-1a), 3.43 (dd, J = 10.0, 6.8 Hz, 1H, H-1b), 2.78 (d, J = 3.6 Hz, 1H, 2-OH), 2.26–2.20 (m, 2H, CH2), 1.40–1.24 (m, 18H, CH2), 0.88 (t, J = 6.8 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 154.3 (C), 138.5 (C), 138.3 (C), 137.9 (CH), 137.3 (C), 128.4 (CH × 2), 128.3 (CH × 2), 128.2 (CH × 2), 128.1 (CH × 2), 127.9 (CH), 127.8 (CH × 2), 127.7 (CH × 2), 127.5 (CH × 2), 123.1 (CH), 110.4 (CH), 73.8 (CH), 73.5 (CH), 73.4 (CH2), 71.2 (CH2), 71.0 (CH2), 70.1 (CH2), 31.9 (CH2), 29.65 (CH2), 29.64 (CH2 × 2), 29.56 (CH2), 29.54 (CH2), 29.4 (CH2), 29.3 (CH2), 29.2 (CH2), 22.7 (CH2), 14.1 (CH3); HRMS (ESI, M + Na+) calcd for C39H52O4Na 607.3758, found 607.3762.
(2R,3S,4Z,6Z)-1,3,5-Tri-O-benzyl-octadec-4,6-diene-1,2,3,5-tetraol (9b). Method A: a solution of hemiacetal 8b (100 mg, 0.19 mmol) and dodecyltriphenylphosphonium bromide (378 mg, 0.74 mmol) in anhydrous tetrahydrofuran (5 mL) was cooled to 0 °C, followed by quick addition of potassium tert-butoxide (83 mg, 0.74 mmol). After stirring for 4 h at this temperature, the reaction mixture was treated with water (2 mL) and extracted with ethyl acetate (3 × 2 mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated. The residue was purified by column chromatography to afford the olefin 9b (98 mg, 90%) as a colorless oil. Method B: a solution of hemiacetal 8c (200 mg, 0.37 mmol) and dodecyltriphenylphosphonium bromide (760 mg, 1.48 mmol) in anhydrous tetrahydrofuran (2 mL) was cooled to 0 °C, followed by quick addition of potassium tert-butoxide (170 mg, 1.48 mmol). After stirring for 2 h at this temperature, the reaction mixture was treated with water (5 mL) and extracted with ethyl acetate (3 × 5 mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated. The residue was purified by column chromatography to afford the olefin 9b (170 mg, 79%) as a colorless oil. Rf 0.55 (EtOAc/Hex = 1/3); [α]23D +15.3 (c 0.5, CH2Cl2); IR (CH2Cl2) ν 3567, 3463, 3064, 3031, 2925, 2854, 1651, 1496, 1457 cm−1; 1H NMR (600 MHz, CDCl3) δ 7.35–7.22 (m, 15H, ArH), 5.76 (d, J = 11.4 Hz, 1H, H-6), 5.77 (dt, J = 12.0, 7.8 Hz, 1H, H-7), 4.87 (d, J = 9.6 Hz, 1H, H-4), 4.80 (s, 2H, CH2Ph), 4.59 (dd, J = 9.6, 4.2 Hz, 1H, H-3), 4.52–4.48 (m, 3H, CH2Ph), 4.29 (d, J = 12.0 Hz, 1H, CH2Ph), 3.92–3.89 (m, 1H, H-2), 3.69 (d, J = 4.8 Hz, 1H, 2-OH), 3.56 (dd, J = 9.6, 4.2 Hz, 1H, H-1a), 3.50 (dd, J = 9.6, 6.0 Hz, 1H, H-1b), 2.35–2.31 (m, 2H, CH2), 1.42–1.40 (m, 2H, CH2), 1.31–1.26 (m, 16H, CH2), 0.88 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (150 MHz, CDCl3) 155.0 (C), 140.3 (C), 139.8 (C), 138.8 (C), 137.6 (CH), 129.1 (CH × 2), 128.9 (CH × 2), 128.8 (CH × 2), 128.7 (CH × 2), 128.5 (CH), 128.3 (CH × 2), 128.2 (CH × 2), 128.0 (CH), 127.8 (CH), 124.6 (CH), 112.1 (CH), 75.3 (CH), 73.6 (CH), 73.5 (CH), 72.7 (CH), 71.9 (CH2), 70.5 (CH2), 32.6 (CH2), 30.34 (CH2), 30.29 (CH2), 30.16 (CH2), 30.11 (CH2), 30.0 (CH2), 29.8 (CH2), 29.4 (CH2), 23.3 (CH2), 14.3 (CH3); HRMS (ESI, M + Na+) calcd for C39H52O4Na 607.3758, found 607.3738.
(2S,3S,4R,5S,6Z)-1,3,4,5-Tetra-O-benzyl-octadec-6-ene-1,2,3,4,5-pentaol (10a). A solution of hemiacetal 8a (100 mg, 0.19 mmol) and dodecyltriphenylphosphonium bromide (567 mg, 1.11 mmol) in anhydrous tetrahydrofuran (1 mL) was cooled to 0 °C, followed by slow addition of lithium hexamethyldisilazane (1 M in tetrahydrofuran, 1.1 mL, 1.11 mmol). After stirring for 24 h at this temperature, the reaction mixture was treated with water (1 mL) and extracted with ethyl acetate (3 × 2 mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated. The residue was purified by column chromatography to afford the olefin 10a (83 mg, 65%, Z/E = 2.4/1) as a colorless oil. 10a-Z: Rf 0.48 (EtOAc/Hex = 1/4); [α]22D −3.5 (c 0.1, CH2Cl2); IR (CH2Cl2) ν 3468, 2925, 2854, 1648, 1496, 1210 cm−1; 1H NMR (600 MHz, CDCl3) δ 7.36–7.23 (m, 18H, ArH), 7.17–7.16 (m, 2H, ArH), 5.65 (dt, J = 10.8, 7.2 Hz, 1H, H-7), 5.44 (dd, J = 11.4, 9.6 Hz, 1H, H-6), 4.79 (d, J = 10.8 Hz, 1H, CH2Ph), 4.74 (d, J = 10.8 Hz, 1H, CH2Ph), 4.63 (d, J = 12.0 Hz, 1H, CH2Ph), 4.48 (d, J = 12.0 Hz, 1H, CH2Ph), 4.44 (dd, J = 9.0, 3.6 Hz, 1H, H-5), 4.41 (d, J = 12.0 Hz, 1H, CH2Ph), 4.40 (d, J = 11.4 Hz, 1H, CH2Ph), 4.37 (d, J = 11.4 Hz, 1H, CH2Ph), 4.31 (d, J = 12.0 Hz, 1H, CH2Ph), 4.10–4.07 (m, 1H, H-2), 3.79–3.76 (m, 2H, H-3, H-4), 3.51 (dd, J = 9.6, 6.6 Hz, 1H, H-1a), 3.47 (dd, J = 9.0, 6.6 Hz, 1H, H-1b), 3.13 (d, J = 4.8 Hz, 1H, 2-OH), 2.06–1.95 (m, 2H, CH2), 1.32–1.25 (m, 18H, CH2), 0.88 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (150 MHz, CDCl3) δ 138.4 (C), 138.22 (C), 138.15 (C), 138.0 (C), 135.5 (CH), 128.3 (CH × 8), 128.1 (CH × 2), 127.99 (CH × 2), 127.97 (CH × 2), 127.7 (CH × 3), 127.6 (CH), 127.53 (CH), 127.50 (CH), 127.0 (CH), 82.5 (CH), 76.6 (CH), 75.4 (CH2), 74.3 (CH), 73.11 (CH2), 73.06 (CH2), 71.2 (CH2), 69.9 (CH2), 69.8 (CH), 31.9 (CH2), 29.7 (CH2 × 3), 29.6 (CH2), 29.5 (CH2), 29.42 (CH2), 29.36 (CH2), 28.1 (CH2), 22.7 (CH2), 14.1 (CH3); HRMS (ESI, M + Na+) calcd for C46H60O5Na 715.4333, found 715.4348. 10a-E: Rf 0.44 (EtOAc/Hex = 1/4); [α]23D −0.1 (c 0.1 CH2Cl2); IR (CH2Cl2) ν 3361, 2925, 2853, 1649, 1497, 1209 cm−1; 1H NMR (600 MHz, CDCl3) δ 7.36–7.25 (m, 18H, ArH), 7.18–7.17 (m, 2H, ArH), 5.70 (dt, J = 15.0, 6.6 Hz, 1H, H-7), 5.40 (dd, J = 15.6, 9.0 Hz, 1H, H-6), 4.78 (d, J = 11.4 Hz, 1H, CH2Ph), 4.75 (d, J = 11.4 Hz, 1H, CH2Ph), 4.62 (d, J = 11.4 Hz, 1H, CH2Ph), 4.47 (d, J = 12.0 Hz, 1H, CH2Ph), 4.43 (d, J = 11.4 Hz, 1H, CH2Ph), 4.39 (d, J = 12.0 Hz, 1H, CH2Ph), 4.35 (d, J = 10.8 Hz, 1H, CH2Ph), 4.33 (d, J = 11.4 Hz, 1H, CH2Ph), 4.11–4.08 (m, 1H, H-2), 4.01 (dd, J = 8.4, 5.4 Hz, 1H, H-5), 3.8 (dd, J = 4.8, 4.8 Hz, 1H, H-4), 3.76–3.75 (m, 1H, H-3), 3.51 (dd, J = 9.0, 6.0 Hz, 1H, H-1a), 3.47 (dd, J = 9.6, 6.6 Hz, 1H, H-1b), 3.19 (d, J = 4.8 Hz, 1H, 2-OH), 2.04–1.98 (m, 2H, CH2), 1.33–1.25 (m, 18H, CH2), 0.88 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (150 MHz, CDCl3) δ 138.5 (C), 138.3 (C), 138.2 (C), 138.0 (C), 136.6 (CH), 128.3 (CH × 8), 128.12 (CH × 2), 128.07 (CH × 2), 127.9 (CH × 2), 127.70 (CH), 127.66 (CH × 2), 127.62 (CH), 127.53 (CH), 127.46 (CH), 127.0 (CH), 82.5 (CH), 80.6 (CH), 76.3 (CH), 75.4 (CH2), 73.1 (CH2), 72.8 (CH2), 71.1 (CH2), 69.9 (CH2), 69.7 (CH), 32.4 (CH2), 31.9 (CH2), 29.70 (CH2), 29.66 (CH2 × 2), 29.5 (CH2), 29.4 (CH2), 29.3 (CH2), 29.0 (CH2), 22.7 (CH2), 14.1 (CH3); HRMS (ESI, M + Na+) calcd for C46H60O5Na 715.4333, found 715.4350.
(2S,3R,4R,5S,6Z)-1,3,4,5-Tetra-O-benzyl-octadec-6-ene-1,2,3,4,5-pentaol (10b). A solution of hemiacetal 8b (100 mg, 0.19 mmol) and dodecyltriphenylphosphonium bromide (567 mg, 1.11 mmol) in anhydrous tetrahydrofuran (1.5 mL) was cooled to 0 °C, followed by slow addition of lithium hexamethyldisilazane (1 M in tetrahydrofuran, 1.11 mL, 1.11 mmol). After stirring for 24 h at this temperature, the reaction mixture was treated with water (1 mL) and extracted with ethyl acetate (3 × 2 mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated. The residue was purified by column chromatography to afford olefin 10b (94 mg, 72%) as a colorless oil. Rf 0.38 (EtOAc/Hex = 1/6); [α]23D +26.5 (c 0.7, CH2Cl2); IR (CH2Cl2) ν 3565, 3475, 3088, 2925, 2854, 1600, 1496, 1457 cm−1; 1H NMR (600 MHz, CDCl3) δ 7.37–7.19 (m, 20H, ArH), 5.67 (dt, J = 10.8, 7.2 Hz, 1H, H-7), 5.47 (dd, J = 11.4, 9.6 Hz, 1H, H-6), 4.80 (d, J = 11.4 Hz, 1H, CH2Ph), 4.74–4.68 (m, 2H, CH2Ph), 4.62 (d, J = 11.4 Hz, 1H, CH2Ph), 4.58–4.46 (m, 4H, CH2Ph, H-5), 4.37 (d, J = 12.0 Hz, 1H, CH2Ph), 4.01 (bs, 1H, H-2), 3.77–3.72 (m, 2H, H-4, H-3), 3.61–3.56 (m, 2H, H-1a, H-1b), 2.98 (d, J = 4.2 Hz, 1H, 2-OH), 2.07–2.01 (m, 2H, CH2), 1.30–1.16 (m, 18H, CH2), 0.88 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (150 MHz, CDCl3) δ 138.45 (C), 138.38 (C), 138.35 (C), 138.1 (C), 135.6 (CH), 128.55 (CH), 128.48 (CH), 128.4 (CH), 128.3 (CH × 7), 128.2 (CH × 2), 127.9 (CH × 2), 127.8 (CH × 2), 127.7 (CH), 127.6 (CH), 127.5 (CH), 127.4 (CH), 127.0 (CH), 126.9 (CH), 81.8 (CH), 78.3 (CH), 74.9 (CH), 74.7 (CH2), 73.3 (CH2), 73.1 (CH2), 71.2 (CH2), 70.5 (CH), 70.2 (CH2), 31.9 (CH2), 29.7 (CH2), 29.6 (CH2 × 2), 29.5 (CH2), 29.4 (CH2), 29.3 (CH2), 28.1 (CH2), 22.7 (CH2), 14.1 (CH3); HRMS (ESI, M + Na+) calcd for C46H60O5Na 715.4333, found 715.4349.
(2S,3R,4R,5R)-1,3,4,5-Tetra-O-benzyl-octadec-6-ene-1,2,3,4,5-pentaol (10c). A solution of hemiacetal 8c (100 mg, 0.18 mmol) and dodecyltriphenylphosphonium bromide (567 mg, 1.11 mmol) in anhydrous tetrahydrofuran (1 mL) was cooled to 0 °C, followed by slow addition of lithium hexamethyldisilazane (1 M in tetrahydrofuran, 1.1 mL, 1.11 mmol). After stirring for 12 h at this temperature, the reaction mixture was treated with water (2 mL) and extracted with ethyl acetate (3 × 2 mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated. The residue was purified by column chromatography to afford olefin 10c (82 mg, 64%) as a colorless oil. Rf 0.68 (EtOAc/Hex = 1/3); [α]23D −7.3 (c 4.8, CH2Cl2); IR (CHCl3) ν 3565, 3475, 3064, 3031, 2925, 2854, 1599, 1495, 1457 cm−1; 1H NMR (600 MHz, CDCl3) δ 7.38–7.18 (m, 20H, ArH), 5.83–5.77 (m, 1H, H-7), 5.54–5.48 (m, 1H, H-6), 4.72–4.04 (m, 9H, CH2Ph, H-5), 4.00 (bs, 1H, H-2), 3.89–3.56 (m, 1H, H-3), 3.84–3.83 (m, 1H, H-4), 3.65–3.55 (m, 2H, H-1), 2.61 (bs, 1H, 2-OH), 2.17–2.05 (m, 2H, CH2), 1.37–1.25 (m, 18H, CH2), 0.89–0.87 (m, 3H, CH3); 13C NMR (150 MHz, CDCl3) δ 138.6 (C), 138.51 (C), 138.47 (C), 138.4 (C), 136.7 (CH), 128.3 (CH × 8), 128.21 (CH), 128.16 (CH), 128.13 (CH), 128.09 (CH), 127.8 (CH), 127.7 (CH × 2), 127.6 (CH × 2), 127.5 (CH), 127.44 (CH), 127.41 (CH), 127.37 (CH), 80.8 (CH), 79.9 (CH), 74.4 (CH2), 74.3 (CH2), 73.93 (CH2), 73.6 (CH), 73.2 (CH2), 71.3 (CH2), 70.07 (CH), 69.4 (CH2), 31.9 (CH2), 29.65 (CH2), 29.62 (CH2), 29.60 (CH2), 29.51 (CH2), 29.47 (CH2), 29.4 (CH2), 29.32 (CH2), 28.1 (CH2), 22.7 (CH2), 14.1 (CH3); HRMS (ESI, M + Na+) calcd for C46H60O5Na 715.4333, found 715.4343.
(2S,3S,4R,5R,6Z)-1,3,4,5-Tetra-O-benzyl-octadec-6-ene-1,2,3,4,5-pentaol (10d). A solution of hemiacetal 8d (168 mg, 0.31 mmol) and dodecyltriphenylphosphonium bromide (954 mg, 1.86 mmol) in anhydrous tetrahydrofuran (1.7 mL) was cooled to 0 °C, followed by slow addition of lithium hexamethyldisilazane (1 M in tetrahydrofuran, 1.9 mL, 1.86 mmol). After stirring for 18 h at this temperature, the reaction mixture was treated with water (2 mL) and extracted with ethyl acetate (3 × 2 mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated. The residue was purified by column chromatography to afford olefin 10d (188 mg, 87%, E/Z = 1/2) as a colorless oil. 10d-Z: Rf 0.65 (EtOAc/Hex = 1/4); [α]26D −39.1 (c 1.0, CH2Cl2); IR (CH2Cl2) ν 3058, 3064, 3031, 2925, 2854, 1496, 1456, 1092, 1066, 1027 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.29–7.22 (m, 20H, ArH), 5.77 (dt, J = 11.2, 7.6 Hz, 1H, H-7), 5.48 (dd, J = 10.4, 10.0 Hz, 1H, H-6), 4.73 (d, J = 11.2 Hz, 1H, CH2Ph), 4.66 (d, J = 11.2 Hz, 1H, CH2Ph), 4.59 (d, J = 11.6 Hz, 1H, CH2Ph), 4.58 (d, J = 11.6 Hz, 1H, CH2Ph), 4.50 (d, J = 11.6 Hz, 1H, CH2Ph), 4.49–4.41 (m, 3H, CH2Ph, H-5), 4.33 (d, J = 11.6 Hz, 1H, CH2Ph), 4.17 (dddd, J = 8.0, 6.0, 6.0, 2.0 Hz, 1H, H-2), 3.90 (dd, J = 4.8, 4.8 Hz, 1H, H-4), 3.80 (dd, J = 4.8, 2.0 Hz, 1H, H-3), 3.54 (d, J = 6.0 Hz, 2H, H-1a, H-1b), 3.44 (d, J = 4.4 Hz, 1H, 2-OH), 2.06–1.88 (m, 2H, CH2), 1.30–1.20 (m, 18H, CH2), 0.88 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 138.3 (C), 138.2 (C), 138.1 (C), 137.9 (C), 136.6 (CH), 128.3 (CH × 8), 128.1 (CH × 2), 127.9 (CH × 2), 127.8 (CH × 2), 127.7 (CH × 2), 127.66 (CH), 127.61 (CH), 127.51 (CH), 127.47 (CH), 126.5 (CH), 81.9 (CH), 77.2 (CH), 74.4 (CH2), 74.1 (CH), 73.2 (CH2), 72.7 (CH2), 71.1 (CH2), 70.1 (CH), 70.0 (CH2), 31.9 (CH2), 29.7 (CH2), 29.63 (CH2 × 2), 29.60 (CH2), 29.5 (CH2), 29.4 (CH2), 29.3 (CH2), 28.2 (CH2), 22.7 (CH2), 14.1 (CH3); HRMS (ESI, M + Na+) calcd for C46H60O5Na 715.4333, found 715.4331. 10d-E: Rf 0.59 (EtOAc/Hex = 1/4); [α]27D −29.6 (c 1.0 CH2Cl2); IR (CH2Cl2) ν 3509, 3061, 3031, 2925, 2854, 1496, 1456, 1209, 1097, 1065, 1027, 973 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.32–7.19 (m, 20H, ArH), 5.63 (dt, J = 15.6, 6.4 Hz, 1H, H-7), 5.51 (dd, J = 15.6, 8.4 Hz, 1H, H-6), 4.80 (d, J = 10.8 Hz, 1H, CH2Ph), 4.66 (d, J = 11.2 Hz, 1H, CH2Ph), 4.59 (d, J = 12.0 Hz, 1H, CH2Ph), 4.56 (d, J = 10.8 Hz, 1H, CH2Ph), 4.51 (d, J = 12.0 Hz, 1H, CH2Ph), 4.47 (d, J = 12.0 Hz, 1H, CH2Ph), 4.44 (d, J = 12.0 Hz, 1H, CH2Ph), 4.32 (d, J = 12.0 Hz, 1H, CH2Ph), 4.16–4.11 (m, 1H, H-2), 4.00 (dd, J = 8.4, 4.8 Hz, 1H, H-5), 3.91 (dd, J = 5.2, 5.2 Hz, 1H, H-4), 3.73 (dd, J = 6.0, 2.0 Hz, 1H, H-3), 3.53 (d, J = 6.4 Hz, 2H, H-1a, H-1b), 3.20 (d, J = 5.6 Hz, 1H, 2-OH), 2.11–2.06 (m, 2H, CH2), 1.40–1.26 (m, 18H, CH2), 0.88 (t, J = 6.8 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 138.4 (C), 138.2 (C), 138.1 (C), 138.0 (C), 137.6 (CH), 128.31 (CH × 2), 128.29 (CH × 4), 128.27 (CH × 2), 128.1 (CH × 2), 128.0 (CH × 2), 127.8 (CH × 2), 127.7 (CH × 4), 127.5 (CH), 127.4 (CH), 126.5 (CH), 81.7 (CH), 80.6 (CH), 77.1 (CH), 74.3 (CH2), 73.2 (CH2), 73.0 (CH2), 71.1 (CH2), 69.9 (CH), 69.8 (CH2), 32.5 (CH2), 31.9 (CH2), 29.7 (CH2), 29.6 (CH2 × 2), 29.5 (CH2), 29.35 (CH2), 29.29 (CH2), 29.20 (CH2), 22.7 (CH2), 14.1 (CH3); HRMS (ESI, M + Na+) calcd for C46H60O5Na 715.4333, found 715.4340.
(2S,3S,4Z,6Z)-2-Hexacosanoylamino-3,5-di-O-benzyl-1-O-(2,3,4,6-tetra-O-benzyl-α-D-galactopyranosyl)-octadec-4,6-dien-1,3,5-triol (13). To a solution of compound 6a (150 mg, 0.15 mmol) and triphenylphosphine (105 mg, 0.40 mmol) in anhydrous THF (2 mL) at 0 °C was added diisopropyl azodicarboxylate (DIAD, 95 μL, 0.43 mmol) followed by dropwise addition of diphenylphosphoryl azide (DPPA, 78 μL, 0.40 mmol) to the reaction flask. After complete addition, the temperature was brought to room temperature by removing ice bath and the reaction was stirred for 1 h. Upon completion of the reaction, the mixture was diluted with EtOAc, and the resulting mixture was washed by water. The organic layer was dried over anhydrous MgSO4, filtered, and concentrated in vacuo. Purification of this residue by column chromatography gives the compound 12 (147 mg, 97%). The compound 12 (686 mg, 0.66 mmol) was added to a round bottom flask followed by the addition of triphenylphosphine (313 mg, 1.19 mmol), pyridine (2.0 mL), water (700 μL), and tetrahydrofuran (7.0 mL). Then, the mixture was warmed up to 60 °C and stirred for 12 h. The solvent was evaporated in vacuum to get a crude amine. This crude amine was dissolved in anhydrous dichloromethane (7.0 mL) at room temperature, 1-[3-(di-methylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDC, 205 mg, 1.07 mmol), hexaeicosanoic acid (306 mg, 0.77 mmol) and HOBt (145 mg, 1.07 mmol) were sequentially added to the solution, and the mixture was continuously stirred for 12 h. The reaction solution was diluted with EtOAc, and the resulting mixture was washed by water. The organic layer was dried over anhydrous MgSO4, filtered, and concentrated in vacuo. This resulting residue was purified by column chromatography to give the amide compound 13 (730 mg, 80%). Rf 0.4 (EtOAc/Hex = 1/5); [α]29D +10.2 (c 1.3, CHCl3); mp = 79 °C; IR (CHCl3) ν 3428, 2918, 2360, 1645, 1454, 1116 cm−1; 1H NMR (600 MHz, CDCl3) δ 7.37–7.19 (m, 30H, ArH), 5.91 (d, J = 9.1 Hz, 1H, NH), 5.78 (d, J = 11.8 Hz, 1H, H-6), 5.73–5.69 (m, 1H, H-7), 4.90 (d, J = 11.4 Hz, 1H, CH2Ph), 4.87 (d, J = 3.6 Hz, 1H, H-1′), 4.79 (d, J = 11.4 Hz, 1H, CH2Ph), 4.73 (d, J = 12.0 Hz, 3H, CH2Ph), 4.69 (d, J = 9.6 Hz, 1H, H-4), 4.65 (d, J = 11.4 Hz, 2H, CH2Ph), 4.61 (dd, J = 8.4, 9.0 Hz, 1H, H-3), 4.55 (d, J = 11.4 Hz, 1H, CH2Ph), 4.45 (d, J = 11.4 Hz, 1H, CH2Ph), 4.372 (d, J = 11.4 Hz, 1H, CH2Ph), 4.367 (d, J = 11.4 Hz, 1H, CH2Ph), 4.18–4.12 (m, 2H, CH2Ph, H-2), 4.04 (dd, J = 9.9, 3.6 Hz, 1H, H-2′), 3.94–3.89 (m, 4H, H-1a, H-3′, H-4′, H-5′), 3.72 (dd, J = 10.8, 3.0 Hz, 1H, H-1b), 3.50–3.43 (m, 2H, H-6a′, H-6b′), 2.22–2.11 (m, 2H, H-8a, H-8b), 1.98–1.86 (m, 2H, CH2), 1.51–1.46 (m, 2H, CH2), 1.34–1.23 (m, 62H, CH2), 0.88 (t, J = 6.6 Hz, 6H, CH3 × 2); 13C NMR (150 MHz, CDCl3) δ 172.6 (C), 153.7 (C), 138.8 (C), 138.7 (C), 138.6 (C), 138.5 (C), 137.8 (C), 137.7 (C), 137.5 (CH), 128.33 (CH × 4), 128.29 (CH × 4), 128.24 (CH), 128.17 (CH × 3), 127.7 (CH), 127.82 (CH × 3), 127.77 (CH × 3), 127.7 (CH), 127.5 (CH × 3), 127.4 (CH × 3), 127.3 (CH × 3), 127.2 (CH), 123.0 (CH), 112.4 (CH), 98.8 (CH), 79.0 (CH), 76.5 (CH), 74.73 (CH), 74.70 (CH2), 73.4 (CH2), 73.0 (CH2), 72.9 (CH2), 72.3 (CH), 70.8 (CH2), 70.3 (CH2), 69.4 (CH), 68.8 (CH2), 68.0 (CH2), 52.3 (CH), 36.7 (CH2), 31.9 (CH2 × 3), 29.7 (CH2 × 20), 29.50 (CH2), 29.45 (CH2), 29.4 (CH2), 29.3 (CH2 × 2), 29.2 (CH2), 25.6 (CH2), 22.7 (CH2 × 3), 14.1 (CH3 × 2); HRMS (ESI, M + H+) calcd for C92H131O9NNa 1416.9716, found 1416.9722.
(2S,3S,5S)-2-Hexacosanoylamino-1-O-(α-d-galactopyranosyl)-octadec-1,3,5-triol (2). Compound 13 (118 mg, 0.08 mmol) was dissolved in MeOH/CHCl3 (3/1 ratio, 2 mL) at room temperature. Pd(OH)2/C (118 mg, Degussa type) was added to the reaction mixture, the reaction vessel was purged with hydrogen, and the mixture was stirred under 60 psi pressure at the same temperature for 1 day. The resulting solution was filtered through celite, the filtrate was concentrated in vacuo, and the residue was purified by flash column chromatography on silica gel to afford the compound 2 (43 mg, 62%). Compound 2 was unable to dissolve in d-solvents such as pure CDCl3, CD3OD or d-DMSO but it was slightly dissolved in d5-pyridine. Rf 0.6 (MeOH/DCM = 1/7); mp = 144–148 °C; [α]24D +39.7 (c 0.3, CHCl3); IR (CHCl3) ν 3280, 2850, 1148, 1454 cm−1; 1H NMR (400 MHz, d5-Pyridine) δ 8.38 (d, J = 8.8 Hz, 1H, NH), 5.39 (d, J = 4.0 Hz, 1H, H-1′), 4.69–4.62 (m, 2H, H-2, H-2′), 4.56 (d, J = 3.3 Hz, 1H, H-4′), 4.50–4.38 (m, 6H, H-3, H-3′, H-5, H-5′, H-6a′, H-6b′), 4.09 (dd, J = 10.4, 5.6 Hz, 1H, H-1a), 3.89 (dd, J = 10.4, 5.6 Hz, 1H, H-1b), 2.69 (t, J = 7.2 Hz, 2H, CH2), 2.45 (t, J = 7.6 Hz, 2H, CH2), 2.41–2.38 (m, 2H, CH2), 2.31–2.04 (m, 4H, CH2), 1.89–1.78 (m, 2H, CH2), 1.65–1.55 (m, 2H, CH2), 1.40–1.23 (m, 60H, CH2), 0.87–0.83 (m, 6H, CH3); 13C NMR (150 MHz, d5-Pyridine 100 °C) δ 173.4 (C), 101.8 (CH), 73.1 (CH), 72.1 (CH2), 71.9 (CH × 2), 71.3 (CH × 2), 70.8 (CH), 63.1 (CH2), 50.0 (CH), 43.2 (CH2 × 2), 39.8 (CH2), 37.1 (CH2), 32.3 (CH2 × 4), 30.1 (CH2 × 13), 30.0 (CH2 × 5), 29.9 (CH2 × 5), 26.9 (CH2), 26.4 (CH2), 24.5 (CH2), 23.0 (CH2 × 3), 14.2 (CH3 × 2). HRMS (ESI, M − H+) calcd for C50H98O9N 856.7236, found 856.7222.
General procedure for ELISA
Supernatant was collected three days after incubation and the production of IL-2 was quantified with DuoSet ELISA Development System (R&D System, MN, USA). Briefly, capture antibody was coated on the plate overnight at 4 °C. The plates were washed and blocked with blocking buffer (1% BSA in PBS). Then, samples were added and incubated for 2 hours at room temperature, washed three times with wash buffer (0.05% Tween 20 in PBS), added the detection antibody, and incubated for 2 hours at room temperature. After washing, the plates were added streptavidin–HRP and incubated for 20 minutes at room temperature. After incubation, the plates were washed, then added substrate solution to each well, and incubated for 20 minutes at room temperature. Finally, added stop solution (2.0 N H2SO4) and determined the optical density (OD450) by using a microplate reader (SpectraMax M2, Molecular device, CA, USA).
Acknowledgements
The authors thank the Ministry of Science and Technology (MOST) in Taiwan (NSC101-2113-M-005-006-MY2) and National Chung Hsing University for financial support. The authors special thank Professor Dr Alice L. Yu and post-doctoral fellow Dr Jung-Tung Hung of Academia Sinica, Taipei 115, Taiwan, for performing the bioassay of analogue 2.
Notes and references
- For reviews on the Wittig reaction, see:
(a) B. E. Maryanoff and A. B. Reitz, Chem. Rev., 1989, 89, 863–927 CrossRef CAS;
(b) E. Vedejs and M. J. Peterson, Top. Stereochem., 1994, 21, 1–157 CAS;
(c) M. Edmonds and A. Abell, in Modern Carbonyl Olefination, ed. T. Takeda, Wiley-VCH, Weinheim, Germany, 2004, ch. 1 Search PubMed;
(d) P. A. Byrne and D. G. Gilheany, Chem. Soc. Rev., 2013, 42, 6670–6696 RSC;
(e) P. T. Parvatkar, P. S. Torney and S. G. Tilve, Curr. Org. Synth., 2013, 10, 288–317 CrossRef CAS.
- R. Kuhn and R. Brossmer, Angew. Chem., 1962, 74, 252–253 CrossRef CAS.
-
(a) G. Wittig and G. Geissler, Justus Liebigs Ann. Chem., 1953, 580, 44–57 CrossRef CAS;
(b) Y. A. Zhdanov, Y. E. Alexeev and V. G. Alexeeva, Adv. Carbohydr. Chem. Biochem., 1972, 27, 277–299 CrossRef.
-
(a) R. Wild and R. Schmidt, Liebigs Ann., 1995, 755–764 CrossRef CAS;
(b) V. Costantino, C. Imperatore, E. Fattorruso and A. Mangoni, Tetrahedron Lett., 2001, 42, 8185–8187 CrossRef CAS;
(c) T. Berkenbusch and R. Bruckner, Chem.–Eur. J., 2004, 10, 1545–1557 CrossRef CAS PubMed;
(d) Y. Niu, X. Cao and X.-S. Ye, Helv. Chim. Acta, 2008, 91, 746–752 CrossRef CAS.
-
(a) V. Aucagne, A. Tatibouet and P. Rollin, Tetrahedron, 2004, 60, 1817–1826 CrossRef CAS PubMed;
(b) M. Cieplak and S. Jarosz, Tetrahedron: Asymmetry, 2011, 22, 1757–1762 CrossRef CAS PubMed.
-
(a) J. R. Pougny, M. M. Nassr and P. Sinay, J. Chem. Soc., Chem. Commun., 1981, 375–376 RSC;
(b) F. Nicotra, F. Ronchetti and G. Russo, J. Org. Chem., 1982, 47, 5381–5382 CrossRef CAS;
(c) P. Allevi, P. Ciuffreda, D. Colomb, D. Monti, G. Speranza and P. Mannito, J. Chem. Soc., Perkin Trans. 1, 1989, 1281–1283 RSC;
(d) F. Nicotra, G. Russo and L. Toma, Tetrahedron Lett., 1984, 25, 5697–5700 CrossRef CAS.
-
(a) T. Natori, Y. Koezuka and T. Higa, Tetrahedron Lett., 1993, 34, 5591–5592 CrossRef CAS;
(b) T. Natori, M. Morita, K. Akimoto and Y. Koezuka, Tetrahedron, 1994, 50, 2771–2784 CrossRef CAS.
-
(a) S. Hong, M. T. Wilson, I. Serizawa, L. Wu, S. Nagendra, O. Naidenko, T. Miura, T. Haba, D. C. Scherer, J. Wie, M. Kronenberg, Y. Koezuka and L. Van Kaer, Nat. Med., 2001, 7, 1052–1056 CrossRef CAS PubMed;
(b) S. Sharif, G. A. Arreaza, P. Zucker, Q.-S. Mi, J. Sondhi, O. V. Naidenko, M. Kronenberg, Y. Koezuka, T. L. Delovitch, J. M. Gombert, M. Leite-De-Moraes, C. Gouarin, R. Zhu, A. Hameg, T. Nakayama, M. Taniguchi, F. Lepault, A. Lehuen, J.-F. Bach and A. Herbelin, Nat. Med., 2001, 7, 1057–1062 CrossRef CAS PubMed;
(c) L. V. Kaer, Nat. Rev. Immunol., 2005, 5, 31–42 CrossRef PubMed.
-
(a) D. G. Pellicci, O. Patel, L. Kjer-Nielsen, S. S. Pang, L. C. K. K. Sullivan, A. G. Brooks, H. H. Reid, S. Gras, I. S. Lucet, R. Koh, M. J. Smyth, T. Mallevaey, J. L. Matsuda, L. Gapin, J. McCluskey, D. I. Godfrey and J. Rossjohn, Immunity, 2009, 31, 47–59 CrossRef CAS PubMed;
(b) D. M. Zajonc, C. Cantu III, J. Mattner, D. Zhou, P. B. Savage, A. Bendelac, I. A. Wilson and L. Tayton, Nat. Immunol., 2005, 6, 810–818 CrossRef CAS PubMed;
(c) M. Koch, V. S. Stronge, D. Shepherd, S. D. Gadola, B. Mathew, G. Ritter, A. R. Fersht, G. S. Besra, R. R. Schmidt, E. Y. Jones and V. Cerundolo, Nat. Immunol., 2005, 6, 819–826 CrossRef CAS PubMed.
- D. J. Baek, J.-H. Seo, C. Lim, J. H. Kim, D. H. Chung, W.-J. Cho, C.-Y. Kang and S. Kim, ACS Med. Chem. Lett., 2011, 2, 544–548 CrossRef CAS PubMed.
- M. Trappeniers, S. Goormans, K. Van Beneden, T. Decruy, B. Linclau, A. Al-Shamkhani, T. Elliott, C. Ottensmeier, J. M. Werner, D. Elewaut and S. Van Calenbergh, ChemMedChem, 2008, 3, 1061–1070 CrossRef CAS PubMed.
-
(a) Y.-F. Yen, S. S. Kulkarni, C.-W. Chang and S.-Y. Luo, Carbohydr. Res., 2013, 368, 35–39 CrossRef CAS PubMed;
(b) R. C. Sawant, J.-T. Hung, H.-L. Chuang, H.-S. Lin, W.-S. Chen, A.-L. Yu and S.-Y. Luo, Eur. J. Org. Chem., 2013, 7611–7623 CrossRef CAS.
- S. S. Kulkarni and J. Gervay-Hague, Org. Lett., 2006, 8, 5765–5768 CrossRef CAS PubMed.
-
(a) K. Worm-Leonhard, K. Larsen and K. J. Jensen, J. Carbohydr. Chem., 2007, 26, 349–368 CrossRef CAS;
(b) M. H. El-Badry, D. Willenbring, D. J. Tantillo and J. Gervay-Hague, J. Org. Chem., 2007, 72, 4663–4672 CrossRef PubMed.
- O. Boutureira, M.-I. Matheu, Y. Díaz and S. Castillón, Carbohydr. Res., 2007, 342, 736–743 CrossRef CAS PubMed.
- I.-S. Kim, S.-J. Kim, J.-K. Lee, Q.-R. Li and Y.-H. Jung, Carbohydr. Res., 2007, 342, 1502–1509 CrossRef CAS PubMed.
- N. Ding, Y.-P. Liu, G.-K. Lu and Y.-X. Li, Chin. J. Chem., 2007, 25, 1069–1071 CrossRef CAS.
- C.-C. Lin, G.-T. Fan and J.-M. Fang, Tetrahedron Lett., 2003, 44, 5281–5283 CrossRef CAS.
-
(a) R. Wagner, J. W. Tilley and K. Lovey, Synthesis, 1990, 785–786 CrossRef CAS;
(b) A. Orsato, E. Barbagallo, B. Costa, S. Olivieri, L. De Gioia, F. Nicotra and B. La Ferla, Eur. J. Org. Chem., 2011, 5012–5019 CrossRef CAS.
- N. A. Borg, S. W. Kwok, L. Kjer-Nielsen, M. C. J. Wilce, D. G. Pellicci, R. Koh, G. S. Besra, M. Bharadwaj, D. I. Godfrey, J. McCluskey and J. Rossjohn, Nature, 2007, 448, 44–49 CrossRef CAS PubMed.
- E. Henon, M. Dauchez, A. Haudrechy and A. Banchet, Tetrahedron, 2008, 64, 9480–9489 CrossRef CAS PubMed.
- H. Iijima, K. Kimura, T. Sakai, A. Uchimura, T. Shimizu, H. Ueno, T. Natori and Y. Koezuka, Bioorg. Med. Chem., 1998, 6, 1905–1910 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra03369h |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.