
Asymmetric aldol reaction organocatalyzed by bifunctional N-prolyl sulfinamides under solvent-free conditions†
Abstract
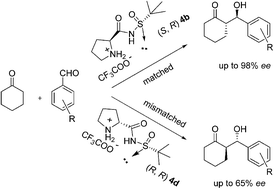
A class of chiral bifunctional N-prolyl sulfinamide and its TFA salts were prepared and proven to be effective for catalyzing the aldol reaction under solvent-free conditions. In general, the corresponding aldol adducts were obtained with high to excellent yields, and satisfactory diastereo-selectivities and enantioselectivities. A matching effect between chiral proline and sulfinamide moieties was observed in the catalysts. The enantioswitching of both enantiomers in the asymmetric aldol synthesis is found to be dominated by the prolyl moiety.
 Please wait while we load your content...
Please wait while we load your content...

