
Liquid–liquid/solid three-phase high-speed counter-current chromatography based on O-carboxymethyl chitosan-functionalized multi-walled carbon nanotubes as solvent additive
Abstract
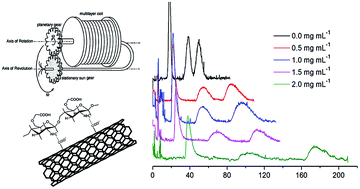
O-Carboxymethyl chitosan-functionalized multi-walled carbon nanotubes (MWCNTs/OMCS) were found to be a novel additive for high-speed counter-current chromatography (HSCCC). It was shown that a kind of liquid–liquid/solid (LLS) three-phase HSCCC system can be established with MWCNTs/OMCS as additive, and in this HSCCC system liquid–liquid partition chromatography and liquid–solid adsorption chromatography were combined. This study revealed that MWCNTs/OMCS can obviously improve peak resolution (Rs). Three flavonol aglycons (quercetin, luteolin and kaempferol) were used as model analytes to study the mechanisms of MWCNTs/OMCS as HSCCC solvent additive to improve Rs. It was indicated that MWCNTs/OMCS improved Rs not by increasing the stationary phase retention but by introducing intermolecular forces: electrostatic interaction, hydrogen bonding interaction and π–π conjugation.
 Please wait while we load your content...
Please wait while we load your content...

