 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient Ru–Fe catalyzed selective hydrogenolysis of carboxylic acids to alcoholic chemicals†
Wenjing
Li
a,
Linmin
Ye
a,
Pei
Long
a,
Jin
Chen
a,
Hiroko
Ariga
b,
Kiyotaka
Asakura
b and
Youzhu
Yuan
*a
aState Key Laboratory of Physical Chemistry of Solid Surfaces, National Engineering Laboratory for Green Chemical Production of Alcohols-Ethers-Esters, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. E-mail: yzyuan@xmu.edu.cn
bCatalysis Research Center, Hokkaido University, Kita-ku N21W10, Sapporo, Hokkaido 001-0021, Japan
First published on 20th June 2014
Abstract
Supported bimetallic Ru–Fe catalysts were prepared using a step-deposition–reduction method. The selective hydrogenolysis of acetic acid to ethanol was investigated as a reaction, which is considered to be related to the transformation of biomass-derived carboxylic acids to fuels and value-added chemicals. An SBA-15-supported Ru–Fe catalyst displayed significant improvements in catalytic performance for the hydrogenolysis of acetic acid to ethanol compared with monometallic catalysts and that with SiO2 as a carrier. When the Ru/Fe atomic ratio was set at 2/1, the prepared catalyst could give a nearly 100% conversion of acetic acid and 88% selectivity to ethanol. The catalyst showed considerable stability in terms of structure and performance for a long-term run on stream. Characterization results indicated that a small portion of Fe species was alloyed with Ru, whereas the other portion of Fe species, likely FeO1+x (0 < x < 0.5), was dispersed on the catalyst surfaces. The Fe species were crucial for the stabilization of Ru–Fe bimetallic nanoparticles and activation of acetic acid molecules in the hydrogenolysis reaction. Moreover, several other carboxylic acids, such as propionic acid, levulinic acid, and lactic acid, could also be efficiently converted to their corresponding alcoholic chemicals or lactone using the optimized Ru–Fe/SBA-15 catalyst under relatively mild conditions.
1. Introduction
The dramatic increase in the consumption of fossil fuels and fossil-derived chemicals has led to environmental problems and exhaustion of energy resources. The efficient utilization of renewable biomass resources at a large scale for producing chemicals and fuels has become a demand for social and industrial development. Most of biomass conversion technology focuses on alcohols, primarily ethanol (EtOH) which is considered as the cleanest liquid fuel alternative to fossil fuel.1,2 Alcohols can further be converted to alkanes with minor losses of energy. In contrast to the very simple but seriously disadvantageous thermochemical pathway of pyrolysis, a more efficient novel way has recently been suggested through carboxylate platforms for the production of biofuels and biochemical from biomass.2,3 In fact, the transformation of lignocellulose to fuel EtOH follows an indirect route involving the fermentation of lignocellulose to acetic acid (AcOH) and hydrogenolysis of AcOH, or direct hydrogenolysis or fermentation of sugars and glucose, or conversion of syngas derived from biomass gasification.3–7 However, the hydrogenolysis of sugars and conversion of biomass-derived syngas only produces low yields of EtOH.5–7 Compared with the direct fermentation of glucose to EtOH (Reaction (1)), the fermentation of glucose to AcOH followed by the hydrogenolysis of AcOH to EtOH can prevent the emission of CO2 (Reaction (2)), achieving 100% carbon yield which against 48% CO2 produced in Reaction (1).8 Thus, the hydrogenolysis of AcOH may be considered as an efficient route that bridges the gap between biomass and fuel EtOH in view of atom economy.
On the other hand, the hydrogenolysis of carboxylic functional group is the crucial reaction for the production of alcohol from the biomass-derived feedstock.9 The development of low-cost, high-efficiency hydrogenolysis routes will open economically viable pathways from renewable resource-derived materials as alternatives to fossil-based chemicals.10,11 Also, the reaction is one of the most useful synthetic tools in the transformation of fats, in particular the synthesis of fatty alcohols from their corresponding carboxylic acids and esters.12,13 Fatty alcohols are nonionic surfactants and are widely used in lubricants, resins, perfumes, cosmetics, shampoos and conditioners.14 Recently, fatty alcohols have been explored for potential use in medicine, health supplements and biofuels.15
However, the hydrogenolysis of short chain carboxylic acids to alcohol in a continuous flow system working in vapor phase under mild conditions seems to be still a problematic step. Further, hydrogenolysis of the C–C bond and over hydrogenation of alcohols may also occur, resulting in cracked products and alkanes, respectively.16–18 The reaction indicates that promoting C–O bond breakage and hydrogenating C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond but suppressing C–C bond cleavage of carboxylic acids will result in high selectivity toward the corresponding alcohols. AcOH can be the model to test the carboxylic acid adsorption and its conversion kinetics because of molecular simplicity and wide range commercial application. Noble metals, such as Pt and Ru-based catalysts, have been intensively explored for the hydrogenolysis reaction of carboxylic acids.16–21 Several researchers have studied the effects of a second metal on the catalysts to improve the activity and selectivity of noble catalysts. For example, Jiang et al. reported that the introduction of Cu to Ru-based catalysts could improve C–O cleavage and suppress C–C cleavage.22 Miyake et al. revealed that the addition of appropriate amount of Sn to Ru-based catalysts promoted both catalytic activity and selectivity in the hydrogenation of fatty acid methyl esters to alcohols.23 Toba et al. reported that Ru–Sn/Al2O3 catalysts demonstrated good yields in the hydrogenolysis of saturated carboxylic acids to their corresponding alcohols,24 but the reported technique required high pressure and temperature.
O bond but suppressing C–C bond cleavage of carboxylic acids will result in high selectivity toward the corresponding alcohols. AcOH can be the model to test the carboxylic acid adsorption and its conversion kinetics because of molecular simplicity and wide range commercial application. Noble metals, such as Pt and Ru-based catalysts, have been intensively explored for the hydrogenolysis reaction of carboxylic acids.16–21 Several researchers have studied the effects of a second metal on the catalysts to improve the activity and selectivity of noble catalysts. For example, Jiang et al. reported that the introduction of Cu to Ru-based catalysts could improve C–O cleavage and suppress C–C cleavage.22 Miyake et al. revealed that the addition of appropriate amount of Sn to Ru-based catalysts promoted both catalytic activity and selectivity in the hydrogenation of fatty acid methyl esters to alcohols.23 Toba et al. reported that Ru–Sn/Al2O3 catalysts demonstrated good yields in the hydrogenolysis of saturated carboxylic acids to their corresponding alcohols,24 but the reported technique required high pressure and temperature.
Ordered mesoporous materials based silicas like MCM-41, SBA-15, and HMS with tunable and regular mesopores, easily accessible internal surface, and relatively high surface areas have been extensively used as carriers for preparing supported metal catalysts. As a result of its large specific surface area, uniform pore size distribution, and better thermal and hydrothermal stability, the catalysts thus prepared are promising for many catalytic reactions in which hydrogen is required, such as photocatalysis, hydrogenation, oxidative dehydrogenation, hydrodesulfurization, and the Fischer–Tropsch synthesis.25–28
In this work, Fe-doped Ru/SBA-15 catalysts (Ru–Fe/SBA-15) prepared by a step-deposition–reduction (SDR) method showed remarkable activity and selectivity for the hydrogenolysis of several bio-derived carboxylic acids, such as AcOH, propionic acid, levulinic acid, butyric acid, and lactic acid to their corresponding alcohols. The catalyst structure was characterized by X-ray fluorescence (XRF), N2 adsorption–desorption isotherms, X-ray diffraction (XRD), H2-temperature-programmed reduction (H2-TPR), AcOH-temperature programmed desorption (AcOH-TPD), transmission electron microscopy (TEM), and X-ray photoelectron spectroscopy (XPS). The relationship between the structure and performance of the bimetallic Ru–Fe/SBA-15 catalysts was further elucidated.
2. Results and discussion
2.1. Effects of Ru/Fe atomic ratio
The catalysts with different Ru/Fe atomic ratios supported on SBA-15 were tested for the hydrogenolysis of AcOH into EtOH at 493 K (Table 1). The reaction yielded products of EtOH, acetaldehyde (AH), ethyl acetate (EtOAc), acetone, and gases (including methane, ethane, and COx). The monometallic catalyst 5% Ru/SBA-15 gave a reasonable AcOH conversion of 15.5% and 74.5% selectivity to methane, indicating that the monometallic Ru catalyst could break the C–C bond in AcOH. The 5% Fe/SBA-15 catalyst had a very low AcOH conversion of 1.3% with 33.1% of selectivity to acetone. However, the Ru–Fe bimetallic catalysts displayed a distinct catalytic performance. When a small amount of Fe was added into Ru/SBA-15 with a Ru/Fe atomic ratio at 15/1, the conversion of AcOH was increased to 35.8% with a 66.8% selectivity to EtOH. The AcOH conversion and EtOH selectivity were gradually increased when the Fe content was further increased. For 5% Ru3–Fe1/SBA-15 as an example, the AcOH conversion increased to 44.0% with 75.2% EtOH selectivity. The AcOH conversion approached a maximal value of 57.2% with a 67.0% selectivity to EtOH when the Fe content was promoted to a Ru/Fe atomic ratio of 2/1. After that, further increase of Fe content caused decreases both in AcOH conversion and EtOH selectivity. The AcOH conversion and EtOH selectivity dropped to 21.7% and 64.9% over the 5% Ru1–Fe1/SBA-15 catalyst, respectively. When the reaction temperature was increased to 533 K, similar trends for the AcOH conversion and EtOH selectivity were observed over the catalysts with different Ru/Fe atomic ratios (Table 2S, ESI†). We found that the highest EtOH yield could be obtained over the 5% Ru2–Fe1/SBA-15 catalyst, although the Ru10–Fe1 and Ru2–Fe1 catalysts performed similar catalytic performance at this temperature.| Catalyst (Ru loading = 5 wt%) | Particle size/nm | Conversion/% | Selectivity/% | TOFc/h−1 | ||||
|---|---|---|---|---|---|---|---|---|
| EtOH | EtOAc | AH | Acetone | Gasesb | ||||
| a Reaction conditions: catalyst weight = 0.20 g, T = 493 K, P(H2) = 3.0 MPa, LHSV(AcOH) = 1.5 h−1, H2/acid = 80. b Gases include methane, ethane, and COx. c TOF was obtained by keeping AcOH conversion below 30% (see Table 1S, ESI for details) and using the data of metal dispersion from H2 adsorption. d ND: not detected. | ||||||||
| Ru/SBA-15 | 18.1 | 15.5 | 22.6 | 1.5 | 1.4 | 0 | 74.5 | 637.1 |
| Ru15–Fe1/SBA-15 | 17.8 | 35.8 | 66.8 | 2.6 | 1.3 | 0 | 29.3 | 827.4 |
| Ru10–Fe1/SBA-15 | 17.1 | 40.5 | 77.3 | 4.5 | 11.6 | 0 | 6.6 | 939.3 |
| Ru3–Fe1/SBA-15 | 16.9 | 44.0 | 75.2 | 5.4 | 12.7 | 0 | 6.7 | 1396.6 |
| Ru2–Fe1/SBA-15 | 16.5 | 57.2 | 67.0 | 5.4 | 20.3 | 0 | 7.3 | 1957.8 |
| Ru1.5–Fe1/SBA-15 | 18.7 | 26.5 | 63.5 | 11.1 | 18.9 | 0 | 6.5 | 1799.8 |
| Ru1–Fe1/SBA-15 | 19.2 | 21.7 | 64.9 | 12.5 | 16.2 | 0 | 6.4 | 1768.6 |
| Fe/SBA-15 | — | 1.3 | 37.5 | 9.6 | 12.2 | 33.1 | 7.6 | 491.9 |
| Ru2–Fe1/SiO2 | 29.6 | 33.1 | 25.4 | 62.3 | 0.5 | 0 | 11.8 | NDd |
| Ru2–Fe1/SBA-15–CoIm-773 K | 29.3 | 24.0 | 63.5 | 23.8 | 3.1 | 0.5 | 9.1 | ND |
| Ru2–Fe1/SBA-15–CoIm-573 K | 3.2 | 57.5 | 76.2 | 1.5 | 0.7 | 0 | 21.6 | ND |
Furthermore, the catalytic activity of the Ru–Fe/SBA-15 bimetallic catalysts underwent a volcano-like tendency with Fe content. The turnover frequency (TOF) was obtained by controlling the AcOH conversion below 30%. A maximum TOF of 1957.8 h−1 was achieved by the Ru2–Fe1/SBA-15 catalyst, which was higher than those obtained with monometallic Ru catalysts in present work and in literature (at 518 K).29 The result indicated that the doped Fe species resulted in a clear promotional effect on the AcOH hydrogenolysis.
2.2. Effects of preparation method and support
The factors affecting the preparation of catalysts, including preparation method and material, had a significant influence on the catalytic performance (Tables 1 and 2). By fixing the Ru/Fe atomic ratio, the 5% Ru2–Fe1/SBA-15–CoIm-773 K catalyst prepared by co-impregnation and calcined at 773 K yielded a 24.0% conversion of AcOH and 63.5% selectivity to EtOH at 493 K, whereas the 5% Ru2–Fe1/SBA-15–CoIm-573 K catalyst prepared by co-impregnation and calcined at 573 K yielded a 57.5% AcOH conversion and 76.2% selectivity to EtOH. The conversion was 68.5% with 67.8% EtOH selectivity over 5% Ru2–Fe1/SBA-15–CoIm-773 K at 533 K. For 5% Ru2–Fe1/SBA-15–CoIm-573 K catalyst, AcOH conversion was up to 99.9% with 99.7% selectivity to gas-phase products (including methane, ethane, and COx) as the temperature increased to 533 K (Fig. 2S, ESI†).| Catalyst (Ru loading = 5 wt%) | Conversion/% | Selectivity/% | ||||
|---|---|---|---|---|---|---|
| EtOH | EtOAc | AH | Acetone | Gasesb | ||
| a Reaction conditions: catalyst weight = 0.20 g, P(H2) = 3.0 MPa, T = 493 K, LHSV(AcOH) = 1.5 h−1, H2/acid = 80. b Gases include methane, ethane, and COx. | ||||||
| Ru2–Fe1/SBA-15 | 57.2 | 67.0 | 5.4 | 20.3 | 0 | 7.3 |
| Ru2–Fe1/ZSM-5 (Si/Al = 50) | 32.9 | 64.6 | 26.6 | 1.2 | 1.4 | 6.2 |
| Ru2–Fe1/ZSM-5 (Si/Al = 25) | 22.5 | 54.9 | 37.4 | 0.6 | 1.1 | 6.0 |
| Ru2–Fe1/HY (Si/Al = 30) | 15.9 | 19.7 | 66.6 | 0.3 | 0 | 13.3 |
| Ru2–Fe1/SiO2 | 33.1 | 25.4 | 62.3 | 0.5 | 0 | 11.8 |
| Ru2–Fe1/Al2O3 | 32.9 | 76.4 | 3.6 | 0.9 | 0.2 | 18.9 |
By replacing SBA-15 with other supports like ZSM-5, HY, SiO2 and Al2O3, the catalysts obtained displayed significantly different catalytic behaviours, giving a lower AcOH conversion in general (Table 2). The conversion of AcOH was 57.2% with 67.0% selectivity to EtOH over 5% Ru2–Fe1/SBA-15 at 493 K. As for the 5% Ru2–Fe1/ZSM-5 (Si/Al = 50) catalyst, the AcOH conversion was 32.9% with 64.6% selectivity to EtOH, while the 5% Ru2–Fe1/ZSM-5 (Si/Al = 25) catalyst displayed a 22.5% AcOH conversion with 54.9% selectivity to EtOH. The 5% Ru2–Fe1/HY yielded a 15.9% AcOH conversion with 19.7% EtOH selectivity and 66.6% EtOAc selectivity. All the catalysts with aluminosilicate zeolites as supports produced considerable EtOAc. The 5% Ru2–Fe1/SiO2 catalyst yielded a 33.1% conversion of AcOH with low selectivity to EtOH (25.4%), but the 5% Ru2–Fe1/Al2O3 catalyst gave a similar AcOH conversion (32.9%) and reasonable EtOH selectivity (76.4%). When the temperature increased to 533 K, AcOH conversion increased but with a higher selectivity to gas-phase products (Fig. 3S, ESI†). The above results suggest that the catalyst performance might have some relationship with the acid–base property of the catalyst supports.
2.3. Effect of reaction temperature
The temperature effects on the hydrogenolysis of AcOH were studied by choosing two typical catalysts, namely, 5% Ru2–Fe1/SBA-15 and 5% Ru2–Fe1/SBA-15–CoIm-573 K. With the 5% Ru2–Fe1/SBA-15 catalyst, the conversion of AcOH and selectivity to EtOH gradually increased as a function of reaction temperature and nearly reached 100% AcOH conversion and 88% selectivity to EtOH at 543 K (Fig. 1). A further increase in the temperature caused a decrease in EtOH selectivity and an increase in gas-phase products (data not shown). With the 5% Ru2–Fe1/SBA-15–CoIm-573 K catalyst, a sharp increase in AcOH conversion was obtained when the reaction temperature increased, but the selectivity to EtOH clearly decreased, accompanying a large amount of gas-phase products.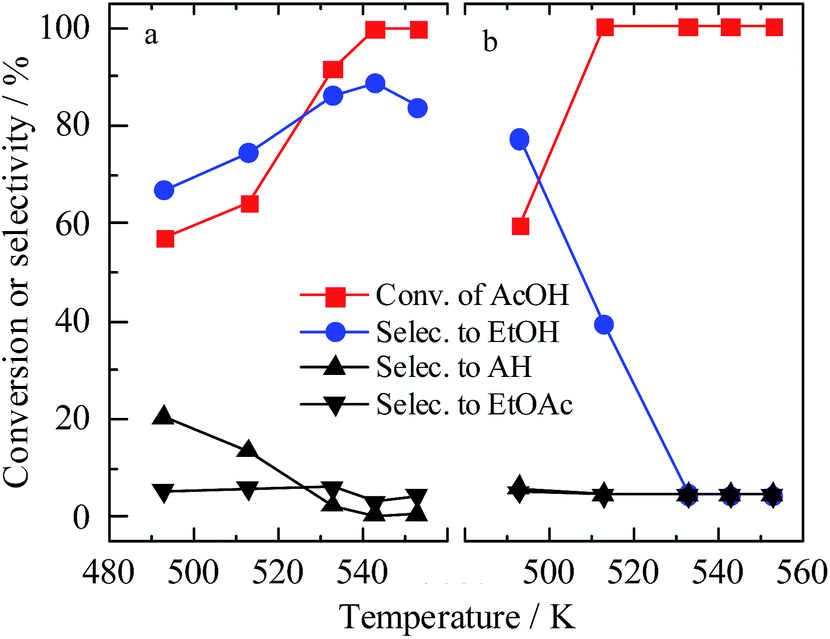 | ||
| Fig. 1 Catalytic performance of 5% Ru2–Fe1/SBA-15 catalysts prepared by different methods: (a) catalyst prepared by SDR method and (b) catalyst prepared by co-impregnation method. | ||
2.4. Hydrogenolysis of several carboxylic acids
Substrates like propionic acid, levulinic acid, butyric acid, and lactic acid were chosen to determine the performance of the optimized 5% Ru2–Fe1/SBA-15 catalyst for the hydrogenolysis of carboxylic acids, except for AcOH (Table 3). The results show an 83.4% conversion of propionic acid and 82.5% selectivity to propanol under conditions similar to the hydrogenolysis of AcOH. In the case of hydrogenolysis of butyric acid, 82.2% conversion was obtained with 85.1% selectivity to butanol. When the 5% Ru2–Fe1/SBA-15 catalyst was used for the hydrogenolysis of lactic acid in aqueous phase, an 80.8% conversion with 76.7% selectivity to 1,2-propandiol as the main product was obtained. The by-products 1-propanol, 2-propanol, and 1,3-propandiol were derived from further hydrogenation or isomerization. For the hydrogenolysis of levulinic acid, a conversion of 85.1% and 85.8% selectivity to γ-valerolactone were obtained. In this case, the CO group was hydrogenated instead of –COOH in levulinic acid possibly because of very low hydrogen pressure (0.75 MPa), and the –OH group generated from CO was esterified with –COOH. When the pressure was increased to 3.0 MPa, the conversion of levulinic acid was up to 95.5% with 53.7% of selectivity to methyltetrahydrofuran (MTHF) which is a product of intramolecular cyclization of 2,5-pentandiol. The results show that the Ru–Fe bimetallic catalyst could perform hydrogenolysis of various carboxylic acids to their corresponding alcoholic chemicals.
| Substrate | Temperature/K | Pressure/MPa | Conversion/% | Selectivitya/% | ||
|---|---|---|---|---|---|---|
| a GVL: γ-valerolactone, MTHF: methyltetrahydrofuran, 1,2-PDO: 1,2-propylene glycol. b Reaction conditions: catalyst weight = 0.1 g, 10 mL of 5% lactic acid aqueous solution was poured into a 50 mL steel autoclave. | ||||||
| Propionic acid | 533 | 3.0 | 83.4 | 82.5 (propanol) | 13.1 (propane) | 4.4 (others) |
| Butyric acid | 533 | 3.0 | 82.2 | 85.1 (butanol) | 12.4 (butane) | 2.5 (others) |
| Levulinic acid | 523 | 0.75 | 85.1 | 85.8 (GVL) | 3.5 (pentanol) | 10.7 (others) |
| Levulinic acid | 543 | 3.0 | 95.5 | 53.7 (MTHF) | 27.5 (pentanol) | 18.8 (others) |
| Lactic acidb | 473 | 3.0 | 80.8 | 76.7 (1,2-PDO) | 13.4 (1,3-PDO) | 9.9 (others) |
2.5. Catalyst stability
The long-term catalytic behaviour of 5% Ru2–Fe1/SBA-15 was investigated under optimized conditions, and the results are shown in Fig. 2. Almost 100% conversion of AcOH was achieved and the selectivity toward EtOH remained about 85% without significant changes for over 300 h. No obvious aggregation of bimetallic particle size and structural collapse of the catalyst occurred after the reaction for 300 h, as proven by the XRD patterns and TEM image (Fig. 1S and 2S, ESI†).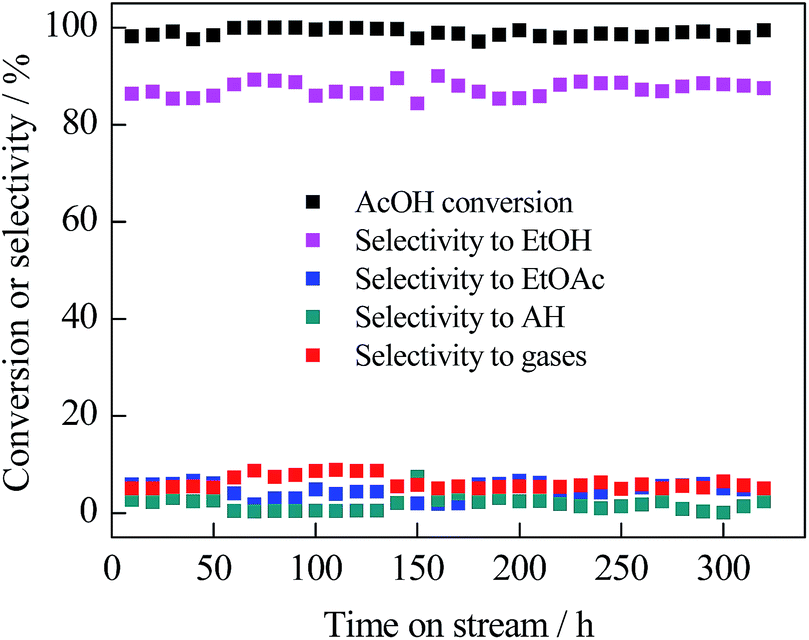 | ||
| Fig. 2 Catalytic performance of 5% Ru2–Fe1/SBA-15 catalyst under the reaction conditions of T = 543 K, P(H2) = 3.0 MPa, LHSV(AcOH) = 1.5 h−1, and H2/AcOH = 80. | ||
2.6. XRF and XRD
The XRF results show that the actual loading amount and Ru/Fe ratios in the monometallic and bimetallic catalysts were highly similar to the theoretical values (Table 4). The XRD patterns of the as-reduced 5% Rux–Fey/SBA-15 catalysts with different Ru/Fe atomic ratios are shown in Fig. 3. The three characteristic peaks at low angles were assigned to the hexagonal mesoporous structure of SBA-15. Bimetal loading of 5% Ru2–Fe1/SBA-15 had no effect on the uniform structure based on the absence of changes at low angles (Fig. 2S, ESI†). Several sharp peaks of monometallic Ru/SBA-15 at 38.4°, 42.1°, 44.0°, 58.3°, 69.3°, 78.3°, and 84.6° were assigned to metallic Ru (100), (002), (101), (102), (110), (103), and (112), respectively. Moreover, the addition of Fe into Ru-based catalysts with different Ru/Fe atomic ratios had no significant effects on peak position and intensity. Monometallic 5% Fe/SBA-15 did not exhibit any characteristic peaks in XRD patterns, indicating a high dispersion of Fe species on SBA-15. However, only three characteristic peaks were observed according to the standard powder XRD data of Fe, and the sharpest peak at 44.6° was close to that of Ru (Fig. 3S, ESI†). Thus, the possible existence of Ru–Fe alloy could not be distinguished based on the presence of a shift in the peak. A similar particle size around 17 nm in the prepared bimetallic catalysts was obtained by the Scherrer equation using the value of half bandwidth in the intensive peak at 44.0°, which agreed with the results of metallic particle size distributions (Fig. 4S, ESI†). Both data were incorporated in Table 4.| Catalyst | M Ru+Fe a/wt% | S BET/m2 g−1 | V pore b/cm3 g−1 | D pore/nm | Average metallic size/nm | H2-chemisorption/cm3 g−1 | |
|---|---|---|---|---|---|---|---|
| By XRDc | By TEM | ||||||
| a Determined by XRF; data in parentheses represent the atomic ratios of Ru/Fe. b Obtained from P/P0 = 0.99. c Calculated by the Scherrer equation. | |||||||
| SBA-15 | — | 661.2 | 0.87 | 5.5 | — | — | 0 |
| Ru/SBA-15 | 5.0 | 390.4 | 0.58 | 6.0 | 17.9 | 18.1 | 0.016 |
| Ru15–Fe1/SBA-15 | 5.0 (13.2/1) | 357.2 | 0.56 | 6.0 | 17.5 | 17.8 | 0.084 |
| Ru10–Fe1/SBA-15 | 5.1 (9.2/1) | 429.7 | 0.62 | 5.7 | 16.3 | 17.1 | 0.091 |
| Ru2–Fe1/SBA-15 | 6.2 (1.7/1) | 496.4 | 0.66 | 5.3 | 16.2 | 16.5 | 0.075 |
| Ru1.5–Fe1/SBA-15 | 6.6 (1.5/1) | 388.3 | 0.52 | 5.1 | 18.9 | 18.7 | 0.064 |
| Ru1–Fe1/SBA-15 | 7.6 (0.9/1) | 348.4 | 0.54 | 5.9 | 19.6 | 19.2 | 0.031 |
| Fe/SBA-15 | 5.2 | 548.7 | 0.62 | 5.4 | — | — | 0 |
 | ||
| Fig. 3 XRD patterns of as-reduced 5% Rux–Fey/SBA-15 catalysts with different Ru/Fe atomic ratios: (a) Ru/SBA-15; (b) Ru10–Fe1/SBA-15; (c) Ru2–Fe1/SBA-15; (d) Ru1–Fe1/SBA-15; and (e) Fe/SBA-15. | ||
2.7. H2-TPR
As shown in Fig. 4, the reduction behaviour of metal oxide was characterized by H2-TPR and H2 consumption was detected by mass spectrometer (m/z = 2). Compared with SBA-15, two peaks appeared at 445 and 570 K in the TPR curve of the as-calcined 5% Ru/SBA-15 sample. The main peak at 445 K was recognized as the reduction of Ru4+ to Ru0,30 and the small peak at 565 K was assigned to the reduction of valent Ru species that strongly interacted with the support, indicating that the Ru oxide was completely reduced to metal Ru after reduction at 623 K. An elevated temperature is needed to reduce Fe oxides, and the peaks at 680 and 840 K are attributed to the reduction of Fe3+ to Fe2+ and Fe2+ to Fe0, respectively.31,32 The Ru–Fe bimetallic samples demonstrated different H2-TPR behaviours compared with monometallic samples, depending on the Ru/Fe atomic ratio. The introduction of non-noble metal with high valence has a negative effect on the ease of reducibility of noble metal oxide.33,34 When a small amount of Fe was added to form the Ru10–Fe1/SBA-15 catalyst, the main peak of H2 consumption shifted to a slightly higher temperature with broadness compared with that in Ru/SBA-15. The increase in Fe amount in Ru/SBA-15 gradually shifted the H2 consumption peak toward higher temperatures. In contrast to the negative effect on the reduction of noble metal oxide caused by the addition of non-noble metal, non-noble metal oxide was reduced at a lower temperature in the presence of a noble metal because of the easy activation of H2 molecules on noble metals.35 Therefore, a reasonable explanation was presented to address the peak broadening at certain Ru/Fe atomic molar ratios of bimetallic catalysts, and an excessive amount of Fe in Ru1–Fe1/SBA-15 led to a small peak at around 616 K. The H2-TPR results demonstrate that particular interactions between Ru and Fe clearly occurred, and a portion of Fe species was reduced under the present conditions. | ||
| Fig. 4 H2-TPR profile of catalysts with different Ru/Fe atomic ratios: (a) Ru1–Fe1/SBA-15; (b) Ru2–Fe1/SBA-15; (c) Ru10–Fe1/SBA-15; (d) Ru/SBA-15; and (e) Fe/SBA-15; (f) SBA-15. | ||
2.8. H2 chemisorption and AcOH-TPD
H2-chemisorption and AcOH-TPD characterizations were applied to obtain more detailed information about the surface chemical properties of the catalysts. As shown in Table 4, SBA-5 was incapable of adsorbing H2 molecules chemically and the Fe/SBA-15 and Ru/SBA-15 displayed relatively poorer absorption of H2 than the Ru–Fe bimetallic catalysts. Increasing contents of Fe in the bimetallic catalysts led to a declining amount of H2 adsorption, indicating that an excessive addition of Fe may have a negative effect on H2 activation.The AcOH-TPD curve of the monometallic Fe/SBA-15 catalyst showed two peaks at 368 and 619 K (Fig. 5). The peak at the lower temperature may be due to the physically adsorbed species, and the peak at the higher temperature may be caused by the chemically adsorbed species.36 For SBA-15, only one peak at lower temperature of 368 K was observed. Similarly, only one peak attributed to physical adsorption appeared in the TPD spectrum of the monometallic Ru/SBA-15 catalyst. Thus, AcOH species were barely absorbed in Ru/SBA-15 or SBA-15 at the temperature performed the AcOH hydrogenolysis reaction (the reaction temperature was higher than 368 K). However, the Ru–Fe bimetallic catalysts displayed a weak desorption peak at 619 K apart from the peak at 368 K. The intensity of the peak assigned to chemical absorption increased with increasing Fe content. This observation suggests that some parts of Fe species dispersed on the bimetallic catalysts may act as adsorption sites for AcOH.
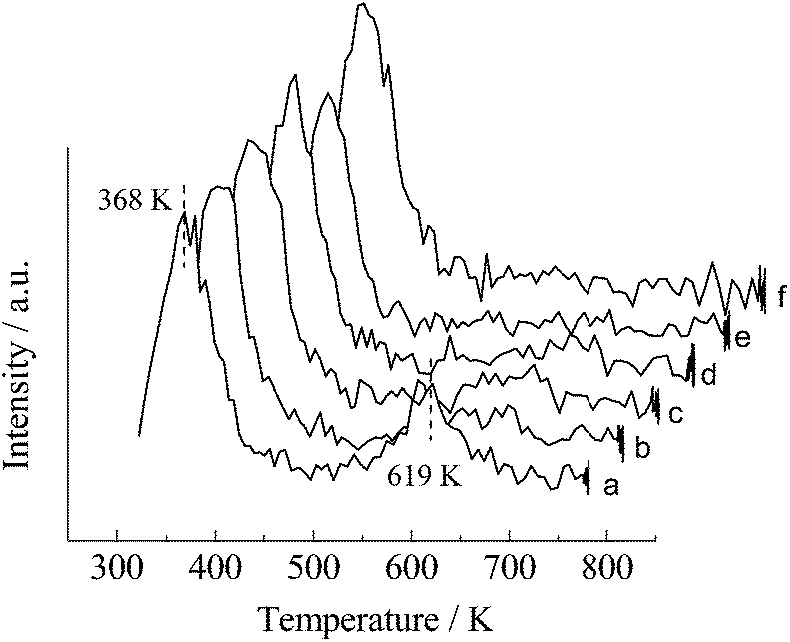 | ||
| Fig. 5 AcOH-TPD result of as-reduced 5% Rux–Fey/SBA-15 catalysts: (a) Fe/SBA-15; (b) Ru1–Fe1/SBA-15; (c) Ru2–Fe1/SBA-15; (d) Ru10–Fe1/SBA-15; and (e) Ru/SBA-15; (f) SBA-15. | ||
2.9. TEM
High-resolution TEM (HRTEM) was used to investigate the inner interaction between Ru and Fe in the bimetallic catalysts at a sub-atomic scale. As shown in Fig. 6a, the HRTEM image of as-reduced Ru2–Fe1/SBA-15 displayed a refined structure of nanoparticles. Intervals of 0.210 and 0.200 nm of the two typical lattice fringes were slightly less than the (002) and (101) lattice spacings in the classic structure of Ru, but the two values were higher than the (110) and (200) lattice spacings of the classic structure of Fe, indicating the doping amount of Fe into metallic Ru. The 56° angle between the two inspected facets was lower than that between the (002) and (101) facets in the classic structure of Ru, indicating the lattice distortion of Ru by doping the second element. The excellent overall dispersions of Ru and Fe particles were detected using STEM-EDX elemental linear scanning. When STEM-EDX elemental mapping was used to investigate the elemental dispersion at a larger scale (Fig. 6d–f), a portion of the Fe species was evenly dispersed on the surface of SBA-15, and the others coherently interacted with Ru domains. In the case of Ru2–Fe1/SiO2, a large average size of bimetallic particles with obvious inhomogeneity was observed (Fig. 5S, ESI†).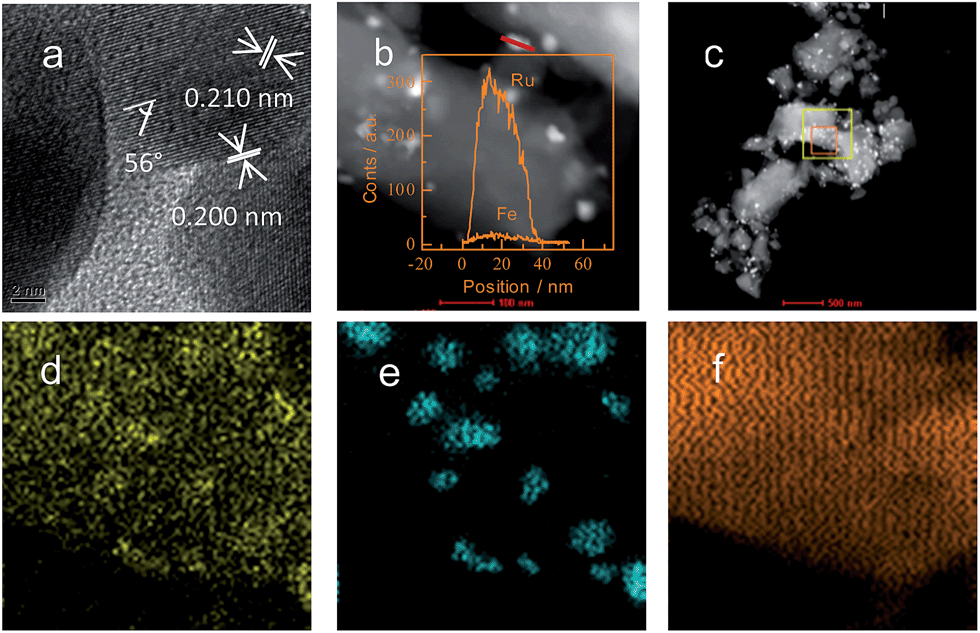 | ||
| Fig. 6 STEM-EDX linear scanning pattern, elemental maps, and HRTEM image of 5% Ru2–Fe1/SBA-15 catalyst: (a) HRTEM image; (b) pattern of STEM-EDX linear scan; (c) dark-field image; (d) STEM-EDX elemental maps for Fe; (e) STEM-EDX elemental maps for Ru; and (f) STEM-EDX elemental maps for Si. | ||
2.10. XPS
XPS measurements of the 5% Ru2–Fe1/SBA-15 and 5% Fe/SBA-15 catalysts were performed to further investigate the elemental valence change in the catalysts. The Fe 2p and Ru 4p XPS data of the Fe/SBA-15 and Ru2–Fe1/SBA-15 catalysts before and after reduction are displayed in Fig. 7, and the deconvolution results are summarized in Table 5. The spectra of the as-calcined and as-reduced Fe/SBA-15 catalysts (Fig. 7a) were similar to each other. The main peak at 711.1 eV and satellite peak at 718.8 eV were attributed to Fe3+ (2p3/2), indicating the presence of Fe3+. These results show that the Fe species on the Fe/SBA-15 catalyst were barely reduced under the present reduction conditions, which agreed with the previous results of H2-TPR. A peak at 711 eV assigned to Fe3+ (2p3/2) also appeared in the as-calcined Ru2–Fe1/SBA-15 catalyst (Fig. 7b). The as-reduced Ru2–Fe1/SBA-15 catalyst exhibited two new peaks distinct from the Fe species on Fe/SBA-15 compared with the as-calcined Ru2–Fe1/SBA-15 catalyst. These peaks at 710.9 and 707.1 eV were characteristic of FeO1+x (0 < x < 0.5) and metallic Fe, respectively.37–39 The concentration of metallic Fe species was estimated to be 10.2% after reduction. The Ru4+ species in Ru2–Fe1/SBA-15 was reduced to metallic Ru at the present condition based on the shift from 462.6 to 461.3 eV (Fig. 7c). This finding supports the previous notion based on H2-TPR results that some Fe species interacting with Ru were reduced to metallic Fe, and others were in the form of FeO1+x after reduction. | ||
| Fig. 7 Fe 2p and Ru 4p XPS of 5% Fe/SBA-15 and 5% Ru2–Fe1/SBA-15 catalysts before and after reduction: (a) Fe 2p spectra of Fe/SBA-15; (b) Fe 2p spectra of Ru2–Fe1/SBA-15; and (c) Ru 4p spectra of Ru2–Fe1/SBA-15. | ||
2.11. Structure-performance correlation and possible catalytic mechanism
The catalytic performance of the 5% Rux–Fey/SBA-15 catalysts, as well as the values of TOF, demonstrates that the addition of Fe considerably promoted AcOH conversion and EtOH selectivity. The consistent relation between the catalytic activity of the 5% Rux–Fey/SBA-15 catalysts and results of H2-chemisorption or AcOH-TPD shows that an appropriate ability to activate H2 molecules and adsorb AcOH molecules was likely the main reason for the increase in AcOH conversion and EtOH selectivity. Multiple characterizations, TEM, XPS, and H2-TPR could explain these phenomena, which are believed to be caused by the synergistic formation of Ru–Fe bimetallic nanoparticles (parts of them in a form of Ru–Fe alloy) and existence of Fe oxide evenly dispersed on the catalyst surfaces. Since Fe catalyst alone showed very low activity for the hydrogenolysis of AcOH either even at higher temperatures, we believe that H2 molecule is activated by the nanoparticles of Ru and Ru–Fe, while the Fe species dispersed on catalysts are functioned to interact with AcOH. However, the H-species activated by Ru catalysts is so active that they are capable of breaking the C–O bonds as well as the C–C bonds, leading to the products mainly in the forms of over hydrogenated or cracked alkanes. In contrast, the H-species activated by Ru–Fe bimetallic nanoparticles shows capability of selectively breaking the C–O bonds in carboxyl group and hydrogenating CO bonds but suppressing C–C bond cleavage, resulting in a markedly enhanced selectivity toward the corresponding alcohol. As we learn from the characterization results above and in literature,34 the proportion of Fe species for the formation of Ru–Fe alloy are very limited under the present pre-treatment condition (pre-treated in 5% H2–95% N2 flow at 623 K for 4 h) and the rest of Fe species in oxide states are dispersed on the catalyst surfaces. Therefore, the catalysts containing Fe species only at an appropriate concentration may exhibit the highest catalytic performance at lower temperatures like 493 K (Table 1). In this case, the catalytic performance is strongly dependent of Ru/Fe atomic ratio. Lower Fe addition brings about limited positive effects. While in the case of excess Fe addition like 5% Ru1.5–Fe1/SBA-15 and 5% Ru1–Fe1/SBA-15 catalysts, the weakness of ability to create active and selective H-species due to the excess of Fe may be the reason for the lower AcOH conversion. According to the present study, when the Ru/Fe atomic ratio was optimized to be 2/1, the 5% Ru2–Fe1/SBA-15 catalyst prepared by SDR method demonstrated the optimal performance in the AcOH hydrogenolysis to EtOH. At an elevated temperature like at 533 K, the catalysts doped with Fe with Ru/Fe ratios from 15/1 to 2/1 can give AcOH conversion approaching to over 90% (Table S2, ESI†) due to the acceleration of mass transfer at higher reaction temperatures (Fig. 1). This is particularly the case for the catalysts of Ru10–Fe1/SBA-15, Ru3–Fe1/SBA-15 and Ru2–Fe1/SBA-15; they give limited differences in performance at 533 K (Table S2, ESI†).
Considering the different performances and structures of the catalysts prepared by SDR and impregnation methods, the functional –NH2 groups had an important function in controlling the final composition and structure of the catalysts. Moreover, the 5% Ru2–Fe1/SBA-15–CoIm-573 K catalyst with smaller bimetallic particle size presents high activity for the C–C bond cleavage with poor selectivity to EtOH production, while the 5% Ru2–Fe1/SBA-15–CoIm-773 K catalyst with larger bimetallic particle sizes shows poor activity. Further, the Ru2–Fe1/SiO2 catalyst showed lower activity and selectivity, although the exact reason for the result remains unclear. However, the larger average size of bimetallic particles with obvious inhomogeneity and lack of uniform channels in 5% Ru2–Fe1/SiO2 compared with those in SBA-15-supported counterpart could not be excluded. Moreover, the surface acidity of the catalysts is believed to have an obvious effect on the selectivity to products. The decrease in Si/Al ratio of aluminosilicate zeolites of ZSM-5 and HY could bring about an increase in selectivity to EtOAc, which most probably is due to the increment of acidity.40 The results demonstrate that the hydrogenolysis of carboxylic acids to corresponding alcohols is a structure-sensitive reaction.
Rachmady and Chen have reported that acyl species is a predominant reaction intermediate which can be hydrogenated to aldehydes and further to alcohols in the process of hydrogenolysis of carboxylic acids.17,41 Combined with the results of hydrogenolysis of several carboxylic acids, a designed mechanism describing the possible pathway for the reaction is shown in Fig. 8. Taking the AcOH hydrogenolysis reaction as an example (R– set as CH3– in Fig. 8), the route of AcOH hydrogenolysis to EtOH proceeds by C–O bond cleavage in AcOH to form CH3CO species, subsequently the hydrogenation of CH3CO species to AH and further hydrogenation to EtOH.17,41 However, C–C cleavage at the first step is possible, which leads to the formation of CH4. A significant amount of CH4 was produced over 5% Ru/SBA-15 because of the excellent capacity of monometallic Ru for cleaving C–C bond. In contrast, the Ru–Fe bimetallic catalysts restricted the occurrence of C–C cleavage, thereby reducing the formation of CH4. The portion of Fe species dispersed on SBA-15 was suspected to interact with AcOH, as shown by AcOH-TPD. The increasing concentration of AcOH on the surface of the catalyst forced the conversion of AcOH. Therefore, the introduction of Fe benefited the hydrogenolysis of AcOH in two aspects. AH was partly produced by hydrogenolysis of AcOH, so a significant amount of AH was detected at relatively low reaction temperature. For instance, the selectivity to AH reached 20.3% using 5% Ru2–Fe1/SBA-15 at 493 K (Fig. 1). AH is highly active and can easily be hydrogenated to EtOH or transformed to EtOAc through self-redox, as shown in the decrease in selectivity of AH to 2.4% at 533 K. The route to EtOAc leads to the decreased selectivity to EtOH. The phenomena of cleaving C–C and C–O of EtOH are also possibly present during the reaction, leading to the formation of CH4 and C2H6. By controlling the experimental conditions, EtOH was cleaved into CH4 with selectivity above 90% on 5% Ru/SBA-15, but the selectivities to CH4 and C2H6 were equally split on 5% Ru2–Fe1/SBA-15 (Fig. 6S, ESI†). This result suggests that the introduction of Fe was advantageous in reducing C–C cleavage.
 | ||
| Fig. 8 Schematic of reactions involved in the conversion of carboxylic acid based on the detected products. | ||
3. Conclusions
In this work, the results of multiple characterizations and catalytic tests show that the introduction of Fe had remarkable effects on the structure and performance of Ru-based supported catalysts. A portion of Fe species interacted with Ru in alloy form, and the other portion dispersed evenly onto SBA-15 at an oxidation state. Fe oxide had an important function in the absorption of AcOH, and Ru–Fe bimetallic nanoparticles exhibited good capacity for activating H2 to reduce AcOH into EtOH. Thus, the Ru–Fe bimetallic catalysts achieved high selectivity toward EtOH by hydrogenolysis of AcOH compared with the monometallic catalysts. Nearly 100% AcOH conversion and 88% EtOH selectivity were achieved by the optimal Ru–Fe/SBA-15 catalyst at an atomic ratio of 2/1 under mild conditions. The Ru2–Fe1/SBA-15 catalyst was highly stable such that high catalytic activity in AcOH hydrogenolysis was maintained over 300 h.4. Experimental
4.1. Chemicals
Chemicals with analytical or guaranteed purities, such as AcOH, propionic acid, levulinic acid, butyric acid, lactic acid, 1,4-dioxane, EtOH, RuCl3·H2O, and Fe(NO3)3·9H2O, were purchased from China Pharmaceutical Group Shanghai Chemical Reagent Co., Ltd. SiO2 was purchased from Qingdao Fine Chemical Co., Ltd. Amphiphilic triblock copolymer (EO)20(PO)70(EO)20 (P123), tetraethyl orthosilicate (TEOS), and 3-aminopropyltriethoxysilane (APTES) were purchased from Sigma-Aldrich. Hydrogen and nitrogen were purchased from Linde Industrial Gases. All the reagents were used as received.4.2. Catalyst preparation
Ordered hexagonal mesoporous silica SBA-15 was synthesized using P123 as the structure-directing agent and TEOS as the silica source according to the detailed procedure in the published literature.42 SBA-15 was functionalized by APTES using the following procedure. Approximately 2.0 g of SBA-15 was suspended in 100 mL of EtOH in a round bottom flask and 5.0 g of APTES was added dropwise. The suspension was refluxed at 363 K for 24 h. Finally, the slurry was filtered, and the solid was washed with EtOH and dried at 333 K overnight. The solid obtained was denoted as NH2–SBA-15.SBA-15-supported Ru–Fe catalysts were prepared by the SDR method according to a similar procedure reported elsewhere,43 which involves two steps, namely, impregnation and reduction. First, 0.1250 g of RuCl3·nH2O was dissolved in 50 mL of distilled water, and the solution was poured into a beaker containing 1.0 g of NH2–SBA-15 with vigorous stirring. Then, 20 mL of NaBH4 aqueous solution (0.079 mol L−1) was added dropwise into the suspension. After 15 min, the mixture was filtered and the solid was washed with distilled water for at least eight times to obtain Ru/NH2–SBA-15. After that, a certain amount of Fe(NO3)3·9H2O was dissolved in 50 mL of distilled water, and Ru/NH2–SBA-15 was re-dispersed in the solution. After the mixture was added dropwise with NaBH4 aqueous solution under vigorous stirring for 1 h, the solid was collected by filtration, washing, drying, and calcination at 773 K for 6 h. The solid was labeled as 5% Rux–Fey/SBA-15. To investigate the influence of additional Fe on the catalytic performance, the Ru loading amount was fixed at 5 wt%, and x/y represented the Ru/Fe atomic ratio. Monometallic Ru/SBA-15 and Fe/SBA-15 catalysts were prepared using the aforementioned SRD method with 0.1250 g RuCl3·nH2O and 3.607 g Fe(NO3)3·9H2O, respectively. In addition, the catalysts with Ru/Fe atomic ratio set to 2/1 were prepared according to the SDR method by replacing SBA-15 with different supports (SiO2, ZSM-5 with different Si/Al ratio for 25 and 50, HY (Si/Al = 30), and Al2O3). The catalysts were denoted as 5% Ru2–Fe1/SiO2, 5% Ru2–Fe1/ZSM-5 (Si/Al = 50), 5% Ru2–Fe1/ZSM-5 (Si/Al = 25), 5% Ru2–Fe1/HY (Si/Al = 30), 5% Ru2–Fe1/Al2O3.
The catalysts labelled as 5% Ru2–Fe1/SBA-15–CoIm-T were prepared for comparison following the procedures described below. First, 1.0 g of non-functionalized SBA-15 was dispersed in 50 mL of acetone solution of RuCl3·nH2O and Fe(NO3)3·9H2O, and the obtained suspension was stirred at room temperature for 12 h. The solvent was evaporated and the solid was dried at 333 K. The solid was calcined at 573 and 773 K for 4 h each, and the obtained catalysts were labeled as 5% Ru2–Fe1/SBA-15–CoIm-573 K and 5% Ru2–Fe1/SBA-15–CoIm-773 K, respectively.
4.3. Catalyst evaluation
The hydrogenolysis of carboxylic acids, such as AcOH, propionic acid, levulinic acid, and butyric acid, was conducted in a stainless tubular fixed-bed reactor equipped with a computer-controlled auto-sampling system. For the hydrogenolysis of levulinic acid, 10 wt% levulinic acid/1,4-dioxane solution was prepared. Typically, 0.2 g of as-prepared catalyst was loaded into a glass tube. The catalyst was pretreated in 5% H2–95% N2 flow (50 mL min−1) at 623 K for 4 h, with a heating rate of 2 K min−1. The temperature was decreased to the target reaction temperature, and pure H2 was introduced into the reactor. The reaction system pressure was precisely controlled by a back-pressure valve to 3.0 MPa (in the case of levulinic acid hydrogenolysis, a pressure of 0.75 MPa was also employed). Carboxylic acid was pumped into the reactor using a Series III digital high-performance liquid chromatography (HPLC) pump (Scientific Systems, Inc.). The outlet stream was injected into a gas chromatograph (GC) equipped with a flame ionization detector and KB-Wax capillary column (30 m × 0.32 mm × 0.33 μm) to detect oxygenates. The products in gas phase were auto-sampled into another GC equipped with a thermal conductivity detector and dual columns, Gaskuropack 54 column (3 m) and active carbon column (2 m), to quantify CH4, CO2, CO, and C2H6. The conversion and product selectivity were calculated using the calibrated area normalization method.The hydrogenolysis of lactic acid was performed in a steel autoclave with a volume of 50 mL using a magnetic stirrer. The reactor was pressurized three times with 1.0 MPa H2 (99.995%) after placing 10 mL of 5% lactic acid aqueous solution and 0.1 g of 5% Ru2–Fe1/SBA-15 in the autoclave. The autoclave was pressurized with the same gas at the desired pressure, and heated to 473 K. After the reaction, the autoclave was cooled to room temperature in a water bath and decompressed. Finally, the liquids and catalysts were separated by decantation. The solution was analysed by HPLC equipped with a refractive index detector and a UV-Vis detector.
4.4. Catalyst characterization
The Ru and Fe loadings on the catalysts were measured by a S8-TIGER XRF spectrometer. Approximately 0.2 g of catalyst and 0.8 g of boric acid were mixed well and compressed to tablets (36 mm diameter and 2 mm thickness). The spectrum was recorded at room temperature.N2 adsorption–desorption isotherms were measured at 77.3 K using a Micromeritics TriStar II 3020 porosimetry analyzer. Prior to adsorption, the sample was degassed under vacuum at 473 K for 2 h. The specific surface area was calculated based on the Brunauer–Emmett–Teller method. According to the Barrett–Joyner–Halenda method, the average pore diameter and pore size distributions were evaluated from the desorption branch of the isotherms.
H2-TPR was performed in a Micromeritics AutoChem II 2920 Chemisorption Analyzer. Samples (0.1 g) were weighed, flushed with 50 mL min−1 high purity He at 473 K for 1 h, and cooled to 323 K. Subsequently, 5% H2–95% Ar flowed through the sample while the temperature increased from 323 to 900 K at a rate of 10 K min−1. Mass spectrometer signals of m/z = 2 were recorded to detect H2 consumption.
Static H2 chemisorption was measured by a Micromeritics ASAP 2020 (M + C) apparatus. The catalyst was pre-reduced by 5% H2–95% N2 flow as the temperature increased from 303 to 623 K at a rate of 2 K min−1 in the pipe furnace. The pre-reduced catalyst was transferred into a quartz test tube in the apparatus, soaked in H2 at 623 K for 15 min, evacuated for 60 min, and then cooled to 308 K. After pretreatment, the catalysts were characterized using a standard program.
AcOH-TPD was performed in a Micromeritics AutoChem II 2920 Chemisorption Analyzer. Approximately 0.2 g of the as-reduced catalyst was sealed in a desiccator, which was full of AcOH vapor for 24 h. The catalyst was loaded into a U-type tube in the analyzer, and swept by Ar as the temperature increased from 323 to 773 K at a rate of 10 K min−1. The desorbed AcOH molecules were detected by a Hiden Qic-20 mass spectrometer with the signals of m/z = 45 and m/z = 43.
XRD patterns were determined using a Philips PANalytical X'pert PRO diffractometer with a graphite monochrometer and Cu Kα radiation (40 kV and 30 mA), and a scanning range from 20 to 90°. According to the Scherrer equation, the particle size of catalysts was calculated using the most intense peak (2θ = 44.0°).
TEM images were taken on a Philips Analytical FEI Tecnai 30 electron microscope operated at an acceleration voltage of 300 V. The sample powder was highly dispersed in EtOH at room temperature, and dropped into copper grids for observation.
XPS measurement was conducted on a PHI QUANTUM 2000 Scanning ESCA Microprobe instrument using an Al Kα radiation source (hv = 1486.6 eV). The XPS spectra of the as-reduced catalysts and as-calcined catalysts were recorded, and the binding energy (BE) was calibrated according to the BE of C1s (284.6 eV).
Acknowledgements
We acknowledge the financial supports from the MOST of China (2011CBA00508), the NSFC (20923004, 21173175, and 21303141), the Research Fund for the Doctoral Program of Higher Education (20110121130002), and the Program for Changjiang Scholars and Innovative Research Team in University (IRT1036).Notes and references
- Y. Lin and S. Tanaka, Appl. Microbiol. Biotechnol., 2006, 69, 627–642 CrossRef CAS PubMed.
- M. T. Holtzapple and C. B. Granda, Appl. Biochem. Biotechnol., 2009, 156, 525–536 CrossRef CAS PubMed.
- H. N. Chang, N.-J. Kim, J. W. Kang and C. M. Jeong, Biotechnol. Bioprocess Eng., 2010, 15, 1–10 CrossRef CAS PubMed.
- V. Pham, M. Holtzapple and M. El-Halwagi, J. Ind. Microbiol. Biotechnol., 2010, 37, 1157–1168 CrossRef CAS PubMed.
- J. J. Spivey and A. Egbebi, Chem. Soc. Rev., 2007, 36, 1514–1528 RSC.
- R. Ooms, M. Dusselier, J. A. Geboers, B. O. de Beeck, R. Verhaeven, E. Gobechiya, J. A. Martens, A. Redl and B. F. Sels, Green Chem., 2014, 16, 695–707 RSC.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- T. Eggeman, D. Verser and E. Weber, in An Indirect Route for Ethanol Production, US Department of Energy: ZeaChem Inc., 2005, DE-FG36–03GO13010 Search PubMed.
- Y. Nakagawa and K. Tomishige, Catal. Surv. Asia, 2011, 15, 111–116 CrossRef CAS.
- S. Harnos, G. Onyestyák and J. Valyon, Appl. Catal., A, 2012, 439, 31–40 CrossRef PubMed.
- J. J. Bozell, L. Moens, D. C. Elliott, Y. Wang, G. G. Neuenscwander, S. W. Fitzpatrick, R. J. Bilski and J. L. Jarnefeld, Resour., Conserv. Recycl., 2000, 28, 227–239 CrossRef.
- E. Uccani, in Heterogeneous Catalysis and Fine Chemicals, ed. M. Guisnet, J. Barrault, C. Bouchoule, D. Duprez, C. Montassier and G. Pérot, Elsevier, Amsterdam, 1988, p. 33 Search PubMed.
- L. M. He, H. Y. Cheng, G. F. Liang, Y. C. Yu and F. Y. Zhao, Appl. Catal., A, 2013, 452, 88–93 CrossRef CAS PubMed.
- L. H. Tan Tai and V. Nardello-Rataj, in Handbook of Detergents Part E: Applications, ed. U. Zoller, CRC press, Taylor and Francis group, Boca Raton, 2009, p. 110 Search PubMed.
- J. L. Hargrove, P. Greenspan and D. K. Hartle, Exp. Biol. Med., 2004, 229, 215–226 CAS.
- W. Rachmady and M. A. Vannice, J. Catal., 2000, 192, 322–334 CrossRef CAS.
- W. Rachmady and M. A. Vannice, J. Catal., 2002, 207, 317–330 CrossRef CAS.
- G. Onyestyák, S. Harnos, S. Klébert, M. Štolcová, A. Kaszonyi and D. Kalló, Appl. Catal., A, 2013, 464, 313–321 CrossRef PubMed.
- J. ten Dam and U. Hanefeld, ChemSusChem, 2011, 4, 1017–1034 CrossRef CAS PubMed.
- C. Luo, S. Wang and H. C. Liu, Angew. Chem., Int. Ed., 2007, 46, 7636–7639 CrossRef CAS PubMed.
- H. J. Wan, R. V. Chaudhari and B. Subramaniam, Energy Fuels, 2012, 27, 487–493 CrossRef.
- T. Jiang, Y. X. Zhou, S. G. Liang, H. Z. Liu and B. X. Han, Green Chem., 2009, 11, 1000–1006 RSC.
- T. Miyake, T. Makino, S.-i. Taniguchi, H. Watanuki, T. Niki, S. Shimizu, Y. Kojima and M. Sano, Appl. Catal., A, 2009, 364, 108–112 CrossRef CAS PubMed.
- M. Toba, S.-i. Tanaka, S.-i. Niwa, F. Mizukami, Z. Koppány, L. Guczi, K.-Y. Cheah and T.-S. Tang, Appl. Catal., A, 1999, 189, 243–250 CrossRef CAS.
- Y. M. Liu, Y. Cao, N. Yi, W. L. Feng, W. L. Dai, S. R. Yan, H. Y. He and K. N. Fan, J. Catal., 2004, 224, 417–428 CrossRef CAS PubMed.
- A. Sampieri, S. Pronier, J. Blanchard, M. Breysse, S. Brunet, K. Fajerwerg, C. Louis and G. Pérot, Catal. Today, 2005, 107, 537–544 CrossRef PubMed.
- A. Martínez, C. López, F. Márquez and I. Díaz, J. Catal., 2003, 220, 486–499 CrossRef.
- J. W. Zheng, H. Q. Lin, Y. N. Wang, X. L. Zheng, X. P. Duan and Y. Z. Yuan, J. Catal., 2013, 297, 110–118 CrossRef CAS PubMed.
- H. Olcay, L. J. Xu, Y. Xu and G. W. Huber, ChemCatChem, 2010, 2, 1420–1424 CrossRef CAS.
- J. L. Li, Y. H. Zhang, Y. X. Zhao, K. Y. Liew and J. P. Hong, Catal. Sci. Technol., 2014, 4, 1005–1011 Search PubMed.
- R. M. M. Abbaslou, A. Tavassoli, J. Soltan and A. K. Dalai, Appl. Catal., A, 2009, 367, 47–52 CrossRef CAS PubMed.
- F. J. Berry, L. W. Lin, C. Y. Wang, R. Y. Tang, S. Zhang and D. B. Liang, J. Chem. Soc., Faraday Trans. 1, 1985, 81, 2293–2305 RSC.
- R. Burch and M. J. Hayes, J. Catal., 1997, 165, 249–261 CrossRef CAS.
- B. D. Li, J. Wang, Y. Z. Yuan, H. Ariga, S. Takakusagi and K. Asakura, ACS Catal., 2011, 1, 1521–1528 CrossRef CAS.
- A. Guerrero-Ruiz, A. Sepúlveda-Escribano and I. Rodriguez-Ramos, Appl. Catal., A, 1992, 81, 81–100 CrossRef CAS.
- J. Fung and I. Wang, Appl. Catal., A, 1998, 166, 327–334 CrossRef CAS.
- C. R. Brundle, T. J. Chuang and K. Wandelt, Surf. Sci., 1977, 68, 459–468 CrossRef CAS.
- N. S. McIntyre and D. G. Zetaruk, Anal. Chem., 1977, 49, 1521–1529 CrossRef CAS.
- V. Schünemann, H. Trevino, W. M. H. Sachtler, K. Fogash and J. A. Dumesic, J. Phys. Chem., 1995, 99, 1317–1321 CrossRef.
- L. Shirazi, E. Jamshidi and M. R. Ghasemi, Cryst. Res. Technol., 2008, 43, 1300–1306 CrossRef CAS.
- L. G. Chen, Y. P. Li, X. H. Zhang, Q. Zhang, T. J. Wang and L. L. Ma, Appl. Catal., A, 2014, 478, 117–128 CrossRef CAS PubMed.
- D. Y. Zhao, Q. S. Huo, J. L. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- Y. N. Wang, X. P. Duan, J. W. Zheng, H. Q. Lin, Y. Z. Yuan, H. Ariga, S. Takakusagi and K. Asakura, Catal. Sci. Technol., 2012, 2, 1637–1639 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The catalytic performance and metal dispersion of as-reduced 5% Rux–Fey/SBA-15 catalysts (Table 1S), catalytic performance of supported 5% Rux–Fey catalysts for AcOH hydrogenolysis at 533 K (Table 2S), catalytic performance of supported 5% Rux–Fey catalysts with different supports for AcOH hydrogenolysis at 533 K (Table 3S), the XRD patterns of 5% Ru2–Fe1/SBA-15 catalyst before and after the catalytic run for 300 h and TEM image after long-term run (Fig. 1S), the low-angle XRD pattern (Fig. 2S), the standard XRD data files of Ru and Fe (Fig. 3S), TEM images and particle size distribution of 5% Rux–Fey/SBA-15 catalysts (Fig. 4S), TEM image and STEM-EDX element mapping of 5% Ru2–Fe1/SiO2 (Fig. 5S), catalytic performance of as-reduced 5% Ru/SBA-15 and 5% Ru2–Fe1/SBA-15 catalysts in EtOH hydrogenolysis at 463 K (Fig. 6S). See DOI: 10.1039/c4ra03201b |
| This journal is © The Royal Society of Chemistry 2014 |