Facile quantitative comparison of specific activities of fusion-tagged enzyme/mutants in cell lysates via prediction of their maximum adsorption by anti-tag antibody immobilized in microplate wells†
Y. L. Li‡
,
X. L. Yang‡,
C. X. He,
X. L. Hu,
J. Pu,
L. Liu,
G. B. Long and
F. Liao*
Unit for Analytical Probes and Protein Biotechnology, Key Laboratory of Clinical Laboratory Diagnostics of the Education Ministry, College of Laboratory Medicine, Chongqing Medical University, Chongqing 400016, China. E-mail: liaofeish@yeah.net; liaofeish@yahoo.com
First published on 19th June 2014
Abstract
The maximum activities of fusion-tagged enzyme/mutants from cell lysates adsorbed by an anti-tag antibody immobilized in microplate wells were predicted to serve as equivalents of their specific activities for comparison with a six-histidine (6His)-tagged esterase and its tagged mutant as models. In brief, (a) a fixed quantity of a monoclonal anti-6His antibody was immobilized in microplate wells; (b) the maximum activity of a tagged enzyme/mutant from a cell lysate for saturation binding to the immobilized antibody (Vs) was predicted from the response of activities of the adsorbed tagged enzyme/mutant to quantities of total proteins in wells from the same lysate; and (c) Vs of tagged enzyme/mutants in lysates served as equivalents of their specific activities for comparison. Prediction of Vs of a tagged enzyme needed initial rates for absorbance changes of over 0.090 in 30 min, the highest occupancy of over 40% of binding sites of the immobilized antibody, and sufficient abundance of the tagged enzyme in lysates. With 0.6 μg antibody for immobilization in wells and total proteins of 10.0 to 128 μg from cell lysates, Vs of the tagged esterase had a coefficient of variation below 10% when the apparent specific activities in lysates varied more than four times. The ratio of Vs of the tagged esterase to the tagged mutant had higher precision and consistency with the ratio of their apparent specific activities from a large number of independent lysates. Hence, Vs predicted for tagged enzyme/mutants in cell lysates was suitable for comparison and may be applicable to verify positive mutants in a library.
Introduction
Enzyme engineering is a research focus of biotechnology. Directed evolution of an enzyme is crucial for enzyme engineering and requires high-throughput (HTP) screening of a mutant library via the assay of enzyme activities in cell lysates after induced expression of candidate mutants in HTP mode.1 However, dynamic efficiency for both cell lysis and induced expression in HTP mode gives many false positive mutants.2 To verify positive mutants, they should be purified to measure their specific activities for comparison against that of the starting enzyme, but the purification process suffers from technical challenges, cost and labor.2b,3 Direct assay of specific activities of mutants in cell lysates is interesting, but no practical methods are available to date. For rational design of mutants of an enzyme or the elucidation of its sequence–activity relationship, it is also preferable to directly and quantitatively compare specific activities of mutants in cell lysates after induced expression. The average of apparent specific activities of a mutant in a large number of independent cell lysates after induced expression can be compared against that of the starting enzyme to judge the difference in their specific activities. However, this comparison inevitably involves heavy cost and labour in the preparation of so many independent cell lysates of each mutant. Hence, new methods are still required for reliable comparison of specific activities of enzyme/mutants in cell lysates after induced expression.To facilitate the purification of recombinant proteins, they are usually expressed via fusion with peptide tags like six-histidine (6His).4 When mutants are fused to a tag, a new approach is available to compare their specific activities in cell lysates after induced expression (Scheme 1). In brief, (a) a fixed quantity of a monoclonal antibody (mcAb) or similar specific adsorbent against the tag is immobilized in microplate wells, (b) a tagged enzyme/mutant from a cell lysate is applied in a well for selective adsorption, (c) the activity of the adsorbed enzyme/mutant is measured, (d) the maximum activity of the tagged enzyme/mutant after saturation binding to the immobilized adsorbent (Vs) is measured, and (e) Vs of each tagged enzyme/mutant as the equivalent of its specific activity can be compared. To generate a mutant library via error-prone PCR or a similar technique, parts of the whole sequence encoding a starting enzyme are spliced for use as templates and peptide tags for fusion expression are always placed on vectors; a mixture of mutated sequences are then linked with the vector for recombinant expression of mutants. As a result, peptide tags for induced expression of a library of enzyme/mutants can be kept unaltered; there should be the same binding ratio of tagged enzyme/mutants to the immobilized adsorbent; the maximum quantities of those tagged enzyme/mutants bound to a fixed quantity of the immobilized adsorbent should be consistent; and Vs can serve as equivalents of their specific activities for comparison as long as nonspecific adsorption is negligible.
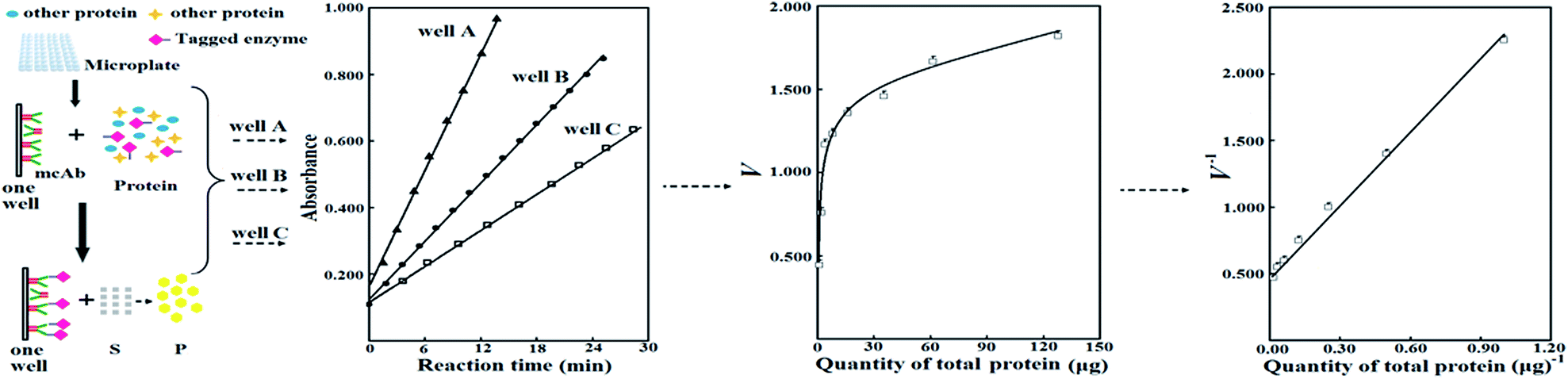 | ||
| Scheme 1 The process for the prediction of Vs. | ||
This direct assay of Vs seems attractive, but is prevented by the following two problems. First, limited affinities of common adsorbents for peptide tags and low abundance of tagged enzyme/mutants in cell lysates result in unsaturated binding to an immobilized adsorbent. Second, there is potential nonspecific adsorption of tagged enzyme/mutants to sites on well surfaces other than the immobilized adsorbent and the competitive nonspecific adsorption of untagged proteins to the immobilized adsorbent. Fortunately, according to chemometrics for reversible adsorption of a tagged enzyme/mutant to an adsorbent immobilized in microplate wells, Vs of the tagged enzyme/mutant may be predicted from the response of activities of the adsorbed tagged enzyme/mutant to quantities of total proteins in wells from the same lysate, at levels of total proteins far below that necessary to achieve saturation binding (Scheme 1).5 The prediction of Vs avoids those challenges associated with direct assay of Vs and may facilitate the comparison of specific activities of tagged enzyme/mutants in cell lysates.
Recently, spectrophotometric simultaneous enzyme-linked immuno-adsorbent assay of two components in one well was reported with attractive advantages,6 but needed a hydrolytic enzyme as the label with high activity in buffers optimal for alkaline phosphatases or glycosidases. A carboxyl esterase active in buffers optimal for glycosidases was expressed via fusion to 6His,7 but its purification faced challenge due to its susceptibility to surfactants. Herein, with an anti-6His antibody as the specific adsorbent, the prediction of Vs was tested with the 6His-tagged esterase and its tagged mutant; results supported that predicted Vs of tagged enzyme/mutants as equivalent of their specific activities was suitable for comparison.
Results and discussion
Chemometrics for the prediction of Vs and optimization of experimental conditions
Let the fixed molar quantity of binding sites on the immobilized mcAb in each well be N, the dissociation constant be K, the total molar quantity of a 6His-tagged enzyme in a well from a sample lysate be n, and the molar quantity of the 6His-tagged enzyme captured in a well from the lysate be b. When equilibrium for the binding is achieved ultimately (Scheme 1), eqn (1) applies:
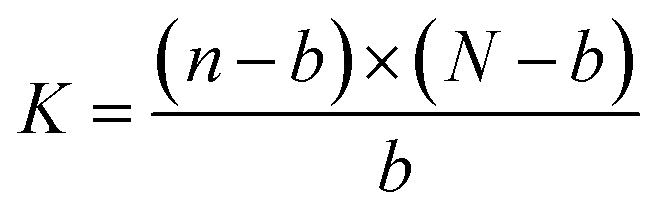 | (1) |
The coefficient of variation (CV) for measuring activities of an enzyme in a lysate is usually about 5%. Hence, with n 20-fold larger than N, eqn (2) applies
 | (2) |
 | (3) |
Letting the abundance of a 6His-tagged enzyme among total proteins in a lysate be P, the quantity of total proteins in a well be T, and the molecular weight of a tagged enzyme be W, eqn (4) applies:
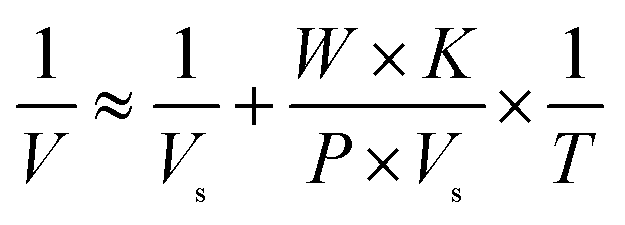 | (4) |
Eqn (4) reflects the saturation binding of a tagged enzyme/mutant as a guest to an mcAb host immobilized in microplate wells, and thus resembles the classical Michaelis–Menten equation of an enzyme for saturation binding of the active site by its substrate. With a tagged enzyme from a cell lysate and a mcAb immobilized in wells, W, P and K are constants. The prediction approach via analysis with eqn (4) thus gives the intercept as the reciprocal of Vs, which as an equivalent of specific activity is suitable for comparison when N is a fixed constant. For confidence, Vs predicted with at least four data and determination coefficient (R2) over 0.95 is accepted. When N is estimated with a tagged enzyme/mutant as the reference, Vs and thus specific activities of other mutants in cell lysates are accessible. Notably, the prediction of Vs does not need K and is resistant to errors in the concentration of total proteins in a sample lysate, but precise dilution of the sample lysate is mandatory.
For reliable prediction of Vs, there should be high quality of initial rates of an adsorbed tagged enzyme/mutant for analysis. In general, high quality of initial rates requires sufficient precision of initial rates, sufficient occupancy percentage of binding sites on the immobilized mcAb and small contribution of nonspecific adsorption to initial rates. For sufficient precision, initial rates for analysis should be over a threshold dependent on random error in absorbance. This threshold was preset for absorbance change of 0.090 in 30 min since random error in absorbance with a microplate reader was about 0.003. As stated above, eqn (4) resembles the Michaelis–Menten equation that describes saturation binding of a substrate to the active site of an enzyme. According to the prerequisites for kinetic analysis of reaction curves of a Michaelis–Menten enzyme,5 the highest initial rate under analysis for reliable prediction of Vs should be generated by an adsorbed tagged enzyme occupying over 40% of binding sites on the immobilized mcAb. The use of any larger T for n much larger than N gives higher occupancy percentage of binding sites on the immobilized mcAb and better precision of initial rates, but inevitably increases potential nonspecific adsorption. The use of a larger quantity of a mcAb in wells for immobilization certainly gives higher initial rates, but reduces the occupancy percentage of binding sites on the immobilized mcAb with the same T. Clearly, with any tagged mutant of higher specific activity, there can be higher initial rates of the adsorbed tagged mutant and enhanced precision of initial rates for analysis under the same conditions. On the other hand, when a tagged enzyme is purified to be in great excess to untagged proteins in a sample, the prediction approach should be easily applied. The abundance of tagged enzyme/mutants among total proteins in cell lysates, quantities of total proteins for adsorption reaction in wells and specific activities of tagged enzyme/mutants are thus primary determinants of the applicability of the prediction approach. Hence, conditions should be optimized carefully to meet all requirements of data quality; the optimal range of quantities of total proteins from lysates for adsorption reaction in wells and the minimum abundance of tagged enzyme/mutants in cell lysates for the applicability of the prediction approach should be carefully examined.
For reliable prediction of Vs, the validation of eqn (2) is mandatory and also requires an optimal range of T with a cell lysate of known abundance of the tagged enzyme/mutant among total proteins and a known quantity of mcAb in wells for immobilization Clearly, a minimum quantity of the mcAb in wells for immobilization to provide initial rates of sufficient quality should be approximated. The absorptivity of 4-nitrophenol with 0.18 mL buffer solution in wells was 8.4 mM−1 cm−1 at pH 7.4. The tagged esterase was assumed to have a minimum specific activity of 150 kU g−1 after purification (S1b, ESI†). With the mcAb from 0.3 to 1.0 μg in wells for immobilization, a sample lysate of the tagged esterase after normal induced expression in 250 mL medium was utilized to estimate values of N (Fig. 1a and b). With the quantities of total proteins in wells large enough for initial rates higher than the aforementioned threshold and occupying more than 50% of binding sites on the immobilized mcAb, N derived from Vs accounted for about 1.8 ng of the tagged esterase with 0.6 μg mcAb in wells for immobilization. Notably, there was a nonlinear response of N (Vs of the same tagged enzyme) to quantities of the mcAb in wells for immobilization (Fig. 1b), indicating dynamic immobilization percentages of the mcAb in wells and a challenge in deriving the minimum quantity of the mcAb in wells for immobilization with the prediction approach. The mcAb used in this report had a nanomolar affinity for 6His tag.8 This affinity suggested the use of moderate quantity of the antibody for the prediction of Vs. When 0.6 μg mcAb was used in wells for immobilization, just about 5% occupancy of binding sites on the immobilized mcAb by the 6His-tagged esterase gave initial rates over the required threshold (Fig. 1a). The upper limit of initial rates accounted for more than 90% occupancy of binding sites on a total of 0.6 μg mcAb for immobilization. When 1.0 μg mcAb was used for immobilization, the achievement of 40% occupancy of binding sites on the immobilized mcAb gave initial rates close to the measurable upper limit. For routine practice, therefore, 0.6 μg mcAb was used in wells for immobilization, unless otherwise stated.
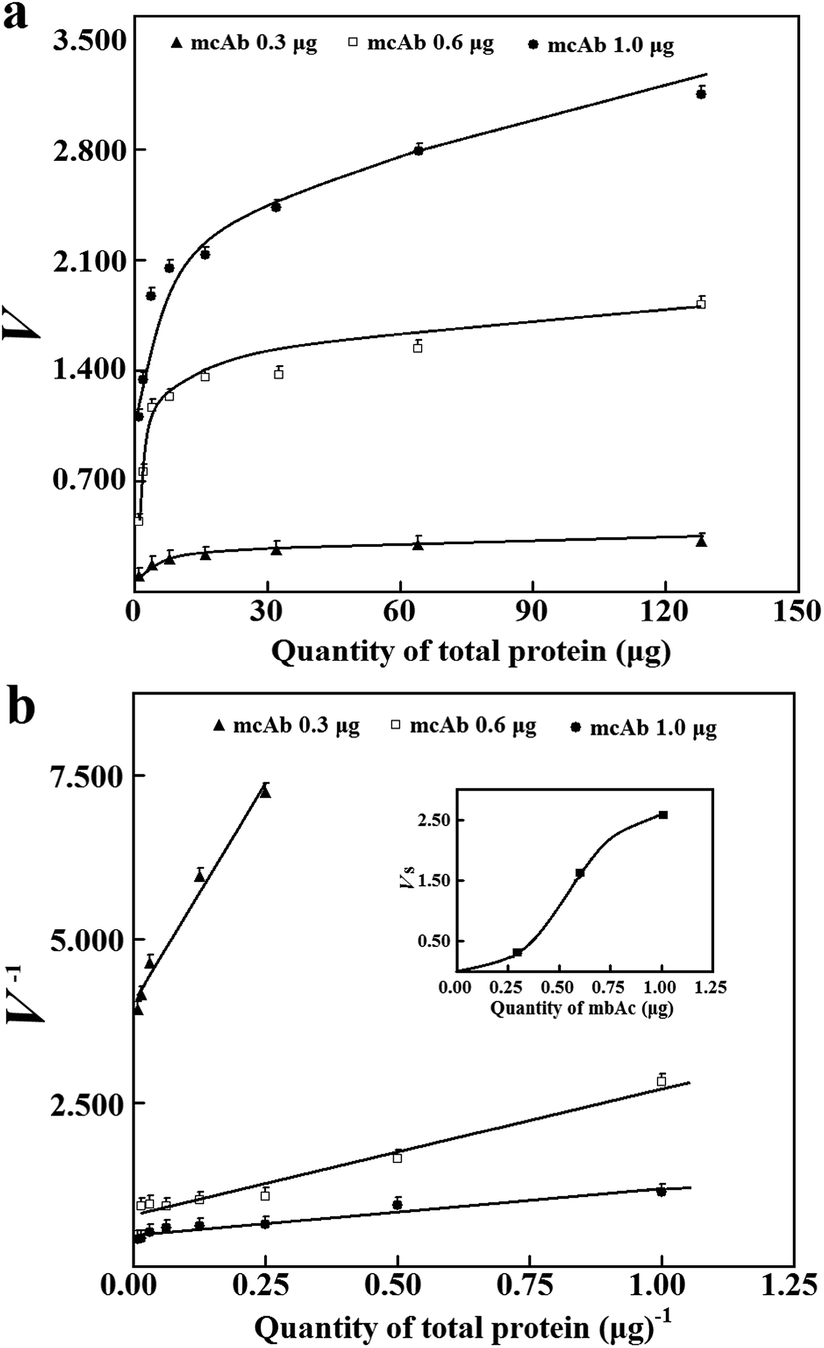 | ||
| Fig. 1 Effects of mcAb quantities on responses of initial rates of the captured esterase. (a) Response of initial rates to quantities of total proteins in wells from a sample lysate with apparent activity of 60 kU g−1. (b) Double-reciprocal prediction of Vs; the inset shows the response of Vs to quantities of the mcAb in wells for immobilization. | ||
With a fixed quantity of the mcAb in wells for immobilization, an optimal range of T with common cell lysates was considered. After induced expression of the tagged esterase in 250 or 4.0 mL medium, the loading of a sample lysate of about 5 μL easily yielded about 10 μg of total proteins in a well (S1c and S1d, ESI†), which can serve as the starting T for the prediction of Vs. In this case, there was already more than 200 ng of a tagged enzyme in a well (S1c and S1d, ESI†), which easily validated eqn (2) when 0.6 μg mcAb was applied in wells for immobilization (Fig. 1b). With just 0.6 μg mcAb in wells for immobilization, the use of 10 μg of total proteins in a well from a sample lysate of the tagged esterase having apparent specific activity of over 8.6 kU g−1 already provided initial rates over the required threshold (Fig. 2a); the use of 128 μg total proteins in a well from such a sample lysate provided occupancy of over 80% of binding sites of the immobilized mcAb (Fig. 2a). However, the use of more sample lysates caused competitive nonspecific adsorption of untagged proteins since initial rates of the adsorbed tagged esterase became smaller (data not given). Hence, an optimal series of T for total proteins varied from 10 to 128 μg, unless otherwise stated, with four or more quantities for adsorption.
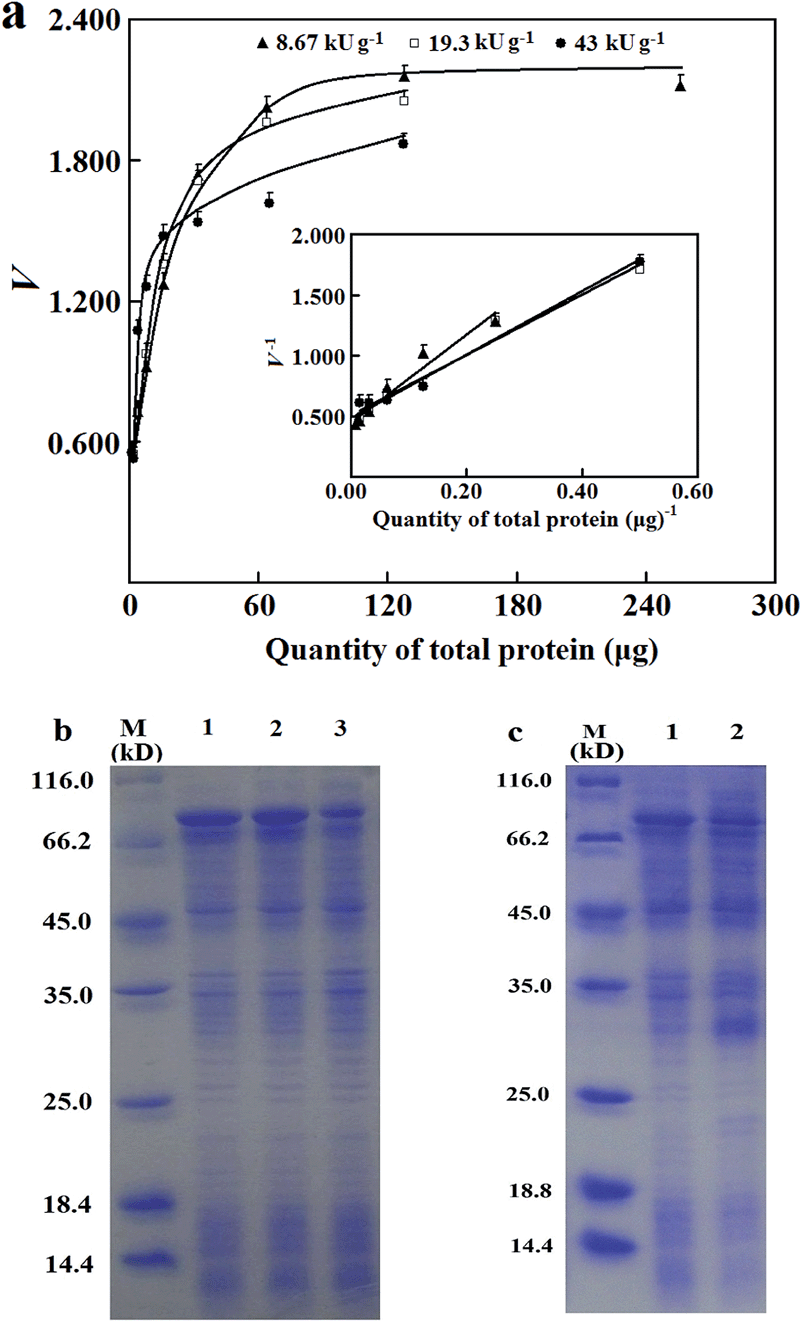 | ||
| Fig. 2 Prediction of Vs of the 6His-tagged esterase in artificial lysates and effects of induction conditions on abundance of the 6His-tagged esterase. (a) The response of initial rates of the 6His-tagged esterase in sample lysates of different abundance (0.60 μg mcAb); the inset is the analysis to predict Vs. (b) SDS-PAGE analysis indicated clear differences in the abundance of the 6His-tagged esterase in those artificial lysates. 1: 43 kU g−1; 2: 19.3 kU g−1; 3: 8.67 kU g−1. (c) SDS-PAGE analysis of abundance of the 6His-tagged esterase in sample lysates prepared after induced expression under different conditions. (1) After induced expression in 250 mL medium; (2) after induced expression in HTP mode in 4.0 mL medium. | ||
The effects of abundance of 6His-tagged esterase in lysates on reliability of Vs were examined to estimate a minimum abundance for applicability of the prediction approach. Artificial lysates were prepared by the dilution of a sample lysate with a control lysate prepared from untransformed bacterial cells. With artificial lysates of the 6His-tagged esterase whose apparent specific activities showed a four-fold difference, Vs showed excellent consistency (Fig. 2a and b). This consistency supported the reliability of the prediction approach and its resistance to variations of abundance of tagged enzyme/mutants in lysates. However, when an artificial lysate was prepared to have an apparent specific activity below 5.4 kU g−1 or abundance of the 6His-tagged esterase below 3.5%, initial rates of the adsorbed 6His-tagged esterase were below the threshold for sufficient precision even with more than 128 μg total proteins for adsorption (S1b, ESI†). This fact indicated competitive nonspecific adsorption of untagged proteins at higher levels of total proteins against the 6His-tagged esterase, and the abundance of over 3.5% of tagged enzyme/mutants among total proteins in lysates may be a threshold for the applicability of the prediction approach. Hence, as a primary determinant of the applicability of the prediction approach, the abundance of 6His-tagged enzyme/mutants in sample lysates after induced expression should be carefully investigated.
Reliability and preliminary applications of Vs
Under the stated conditions to measure initial rates of the adsorbed 6His-tagged esterase, the saturation of the immobilized mcAb was easily observed when 0.6 μg mcAb was used in wells for immobilization (Fig. 1a). Analyses of initial rates with absorbance changes of over 0.090 in 30 min according to eqn (4) gave consistent Vs with R2 over 0.95 while the analyses of smaller initial rates gave Vs having larger deviations and smaller R2 (Fig. 1b). Only when the highest initial rate among data being analysed accounted for >40% occupancy of binding sites on the immobilized mcAb was there consistent Vs from the same response curve. Vs of the tagged esterase from the same sample lysate under optimized conditions had CV below 9% (n = 5), supporting high precision of Vs. Thus, when all prerequisites were met, the prediction approach was applicable.To accelerate comparison, induced expression of tagged mutants in HTP mode in a small volume of medium was always preferred. Notably, after induced expression in 4.0 mL medium, the abundance of the 6His-tagged esterase was clearly lower than that after induced expression in 250 mL medium (Fig. 2c). The applicability of the prediction approach was examined for sample lysates of 6His-tagged esterase after induced expression in 4.0 mL medium. After induced expression of the 6His-tagged esterase in 4.0 mL medium, apparent specific activities in 120 sample lysates exhibited the largest difference of 500% and CV of 25% (Table 1; Fig. 3b; S1c, ESI†). When the 6His-tagged esterase was directly induced in a 96-well microplate, apparent specific activities in 96 sample lysates exhibited the largest difference of 10-fold and CV of about 65% (data not given). These facts should account for many false positive mutants after HTP screening of a mutant library and support the necessity of verification of such positive mutants in the library. Moreover, in sample lysates of the tagged esterase after induced expression in 4.0 mL medium, the abundance of the 6His-tagged esterase had an average of about 5%, but the minimum abundance of the 6His-tagged esterase was just about 2.1% at 99% confidence limit (Fig. 3a and b). Nearly 20% of sample lysates of the 6His-tagged esterase after induced expression in 4.0 mL medium had abundance below 3.5% (Fig. 3b). Thus, for universal applicability of the prediction approach, sample lysates after induced expression in 4.0 mL medium did not always meet the prerequisites.
| Sample | Induced expression in 4 mL medium | Induced expression in 250 mL medium | |||||
|---|---|---|---|---|---|---|---|
| Total proteins (g L−1) | Apparent specific activity (kU g−1) | Tagged protein abundance (%) | Total proteins (g L−1) | Apparent specific activity (kU g−1) | Tagged protein abundance (%) | Vs | |
| a Numbers in parentheses indicate assays with independent sample lysates. Activity was absorbance change in 30 min.b Indicates no significant difference from the approximated ratios of apparent specific activities in a large number of independent sample lysates.c Each time with a pair of sample lysates with the same microplate. | |||||||
| Esterase | 6.3 ± 1.2 (120) | 6.9 ± 1.7 (120) | 5.0 ± 1.0 (120) | 6.0 ± 1.6 (25) | 50.5 ± 7.6 (25) | 34 ± 5 (25) | 1.7 ± 0.2 (5) |
| Mutant | 6.1 ± 0.6 (35) | 2.2 ± 0.7 (35) | 4.2 ± 1.0 (35) | 7.2 ± 1.4 (11) | 17.0 ± 1.7 (11) | 34 ± 3 (11) | 0.62 ± 0.04 (6) |
| Ratio | 3.2 ± 0.8 | 3.0 ± 1.0 | 2.8 ± 0.2 (4)b,c | ||||
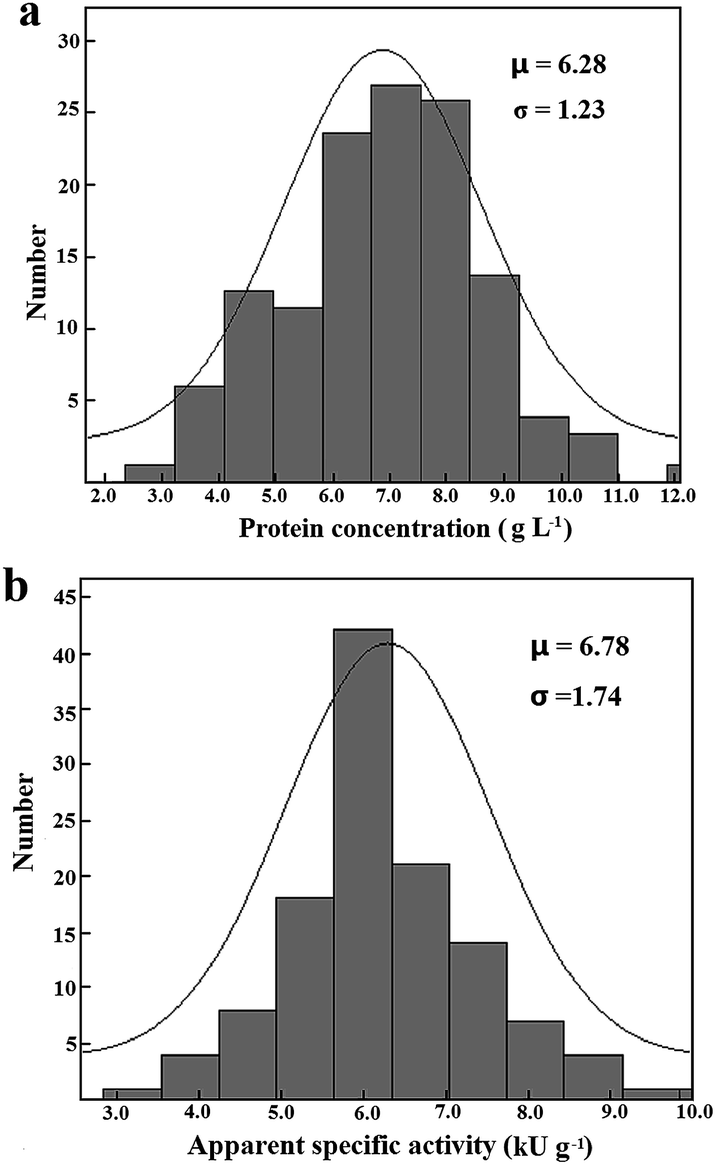 | ||
| Fig. 3 Distribution of concentrations of total proteins (a) and apparent specific activities (b) of the 6His-tagged esterase in a total of 120 cell lysates prepared in HTP mode in 4.0 mL medium. | ||
The applicability of the prediction approach was further examined for sample lysates of 6His-tagged esterase after induced expression in 250 mL medium. After induced expression of the tagged esterase in 250 mL medium, the apparent specific activity had CV of about 15% (n = 25; S1d, ESI†), and was about 7.4-fold that after induced expression in 4.0 mL medium. The average of the abundance of the 6His-tagged esterase was about 30% after induced expression in 250 mL medium; this abundance was comparable to that of a bacterial uricase after induced expression under similar conditions.3 In sample lysates after induced expression in 250 mL medium, the minimum abundance of tagged enzyme/mutants may easily meet the threshold of 3.5% at 99% confidence. More importantly, with sample lysates of the 6His-tagged esterase having abundance of over 6% or apparent specific activities higher than 10 kU g−1, there were consistent Vs values (Fig. 4a). Thus, the prediction approach was applicable to sample lysates when the abundance of tagged enzyme/mutants among total proteins met the requirement.
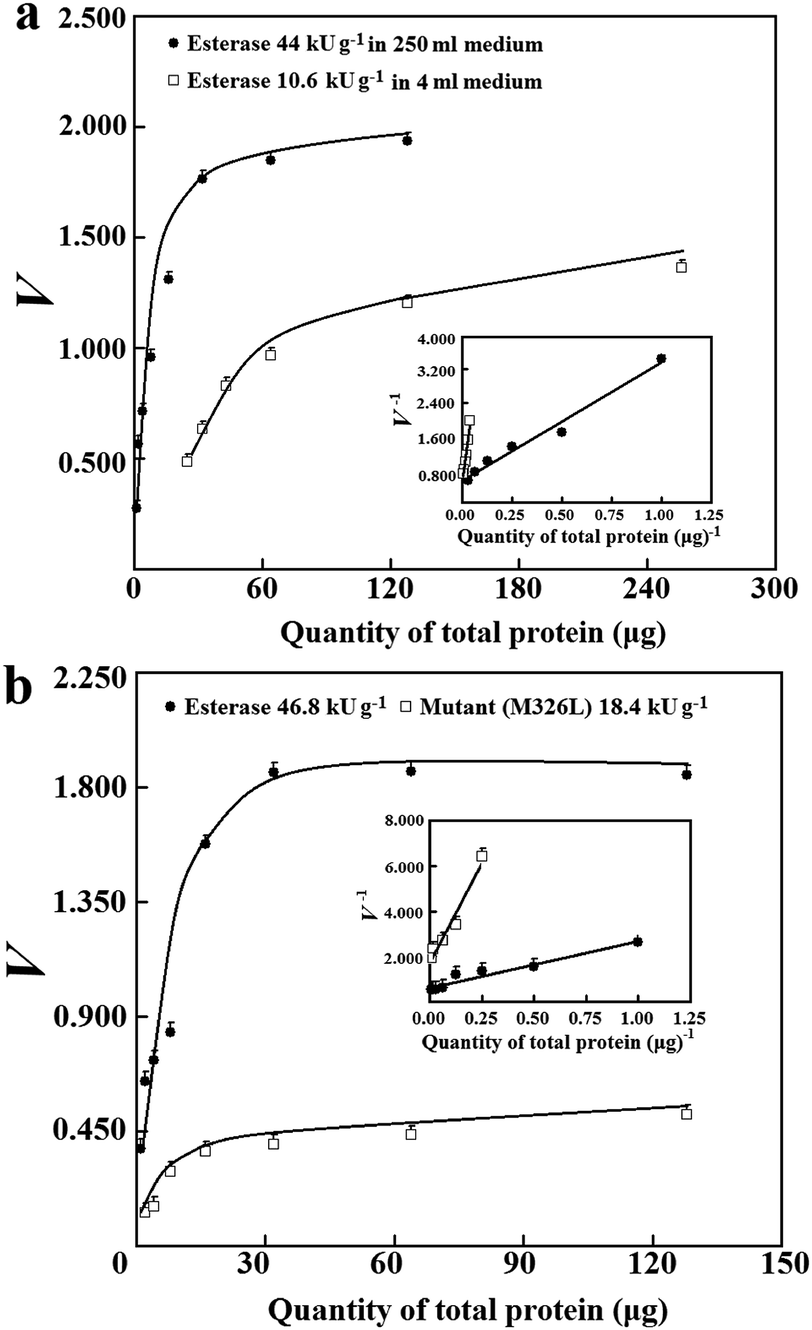 | ||
| Fig. 4 Application of the prediction approach. (a) The prediction of Vs of the 6His-tagged esterase in sample lysates after induced expression in 250 mL medium and 4.0 mL medium (0.60 μg mcAb). (b) Comparison of Vs of the 6His-tagged esterase and the tagged mutant (M326L) after they were induced for expression in 250 mL medium. | ||
The prediction of Vs was tested for estimating a ratio between specific activities of two 6His-tagged enzymes. The 6His-tagged esterase was sensitive to trace quantities of common surfactants except for Tween-20 below 0.07%; its specific activity after affinity chromatography through Ni2+–NTA was smaller than its apparent specific activity in lysates after induced expression in 250 mL medium (S1b, ESI†); the use of other peptide tags presented similar challenges.7 With sample lysates after induced expression in 4.0 mL medium, the ratio of apparent specific activities of the 6His-tagged esterase and its 6His-tagged mutant was 3.2 ± 0.8 considering error propagation (Table 1; S1c, ESI†). Similar to the 6His-tagged esterase, there were about 7.8-fold higher apparent specific activities of the 6His-tagged mutant after induced expression in 250 mL medium in comparison to those after induced expression in 4.0 mL medium; sample lysates of the mutant after induced expression in 4.0 mL medium were unsuitable for prediction of Vs (S1e, ESI†). With a large number of lysates of the 6His-tagged esterase and mutant after induced expression in 250 mL medium, the ratio of their apparent specific activities was 3.0 ± 1.0 considering error propagation (Table 1; S1d, ESI†). However, the ratio of their Vs predicted via analysis with eqn (4) was 2.8 ± 0.2 (Table 1; Fig. 4b). Clearly, the ratio of Vs predicted via eqn (4) showed much enhanced precision and was consistent with the approximated ratio of apparent specific activities for the same pair of 6His-tagged enzymes in a large number of independent sample lysates after induced expression in 250 mL or 4.0 mL medium (Table 1). Hence, the prediction approach was suitable for comparison of specific activities of tagged enzyme/mutants after normal induced expression.
Higher precision of the ratio of Vs of two tagged enzymes should greatly facilitate the comparison of specific activities and even cross-validation of positive mutants after screening of a mutant library.3 6His-tag was widely used for recombinant expression of enzymes.4 The tagged esterase was susceptible to surfactants needed for regeneration of the affinity matrix (S1b, ESI†). The use of mcAbs to capture tagged enzymes usually caused no interference with the assay of enzyme activities and benefited the elucidation of their sequence–activity relationship. Hence, the prediction of Vs of tagged enzyme/mutants for saturation binding to a fixed quantity of a specific adsorbent immobilized in microplate wells was promising for comparison of specific activities of tagged enzyme/mutants in cell lysates.
Conclusions
With an optimized quantity of a specific adsorbent like a mcAb against a fusion tag for immobilization in wells, the maximum activity of a tagged enzyme after saturation binding to the immobilized adsorbent was predicted via analysis of the response of initial rates of the captured tagged enzyme to quantities of total proteins in wells from the same lysate. By the prediction of Vs, equivalents of specific activities of a tagged starting enzyme and tagged mutants in lysates were obtained and found to be suitable for comparison; this prediction approach may be applicable to cross-validate positive mutants after screening of a library of tagged mutants in lysates and to rapidly elucidate sequence–activity relationships of an enzyme and its mutants.Materials and methods
Chemicals and materials
Mouse monoclonal antibody against 6His-tag (mcAb) was from Nanjing Zhongding Biotechnology Co. Ltd (Nanjing, China; catalogue no. ABM-00114). Cultivation media, cell strains, bovine serum albumin (BSA) and isopropanyl-β-D-thiogalactoside (IPTG) were from Shanghai Sangon Biotechnology Co. Ltd (Shanghai, China). 4-Nitrophenyl butyrate was from Qingdao Vochem Co. Ltd, China (Qingdao, Shandong, China). Other chemicals were from Aladdin Reagents Co. Ltd (Shanghai, China).The coding sequence of the carboxyl esterase was synthesized according to its reported sequence (gi: 29893336).7 Synthesis of the coding sequence and site-directed mutagenesis were performed by Beijing Taihe Biotechnology Co. Ltd (http://www.taihegene.com/; S1a in ESI†). Coding sequences were inserted into pET29a vectors.
Conventional assay of initial rates of esterase
Tris–HCl buffer at 50.0 mM and pH 7.4 was pre-incubated at 25 ± 0.5 °C before use for activity assay throughout the work. 4-Nitrophenyl butyrate as the substrate was dissolved in methanol at no less than 25 mM and diluted with the Tris–HCl buffer just before use. For routine assay of activity, absorbance at 405 nm was recorded at 20 s intervals with a MAPADA UV 1600 PC spectrophotometer. Each reaction mixture of 1.0 mL contained 50 μL sample and 950 μL substrate solution for a final concentration of 0.25 mM. Reaction was initiated by the addition of a sample. There was a linear increase in absorbance until 0.890. Spontaneous hydrolysis increased absorbance at 405 nm to about 0.06 in 30 min and was corrected with an appropriate control. One unit of esterase activity released 1 μmol of 4-nitrophenol per minute.Induced expression and preparation of sample lysates
The lysis buffer used throughout the work was 20 mM Tris–HCl at pH 7.4.For normal induced expression, transformed Escherichia coli BL21 (DE3) cells were induced in 250 mL TB medium. After induction with 1.0 mM IPTG for 20 h at 18 °C, transformed cells collected via centrifugation were lysed in 25 mL buffer by sonication treatment at 4 °C for a total of 50.0 min, via continuous treatment for 3 s at 5.0 s intervals with 28% amplitude (SONICS Uibra Cell, Sonics & Materials, Inc., Newtown, CT, USA). The supernatant after centrifugation at 4500 × g for 20 min at 4 °C was a sample lysate after normal induced expression. Cells transformed with a blank pET29a vector were processed in the same way to yield a control lysate with total proteins >7.0 g L−1 but negligible esterase activity (<3 U g−1). Each sample lysate was diluted by more than 6-fold with a control lysate before loading into wells (final concentrations of total proteins after dilution were over 6.0 g L−1). Artificial lysates were prepared by the dilution of a sample lysate after normal induced expression with the control lysate. This dilution process reduced nonspecific adsorption of the tagged esterase to below 10%, while the use of the lysis buffer or BSA for dilution led to >30% nonspecific adsorption.
For induced expression in HTP mode, transformed cells were amplified in 4.0 mL TB medium in small glass flasks in HTP mode, and then induced with final 1.0 mM IPTG for 20 h at 18 °C. Afterwards, cells were collected via centrifugation and lysed in a total of 1.0 mL buffer, via continuous sonication treatment by the same instrument as mentioned above at 4 °C for 20.0 min by using treatment for 3.0 s at 5.0 s intervals with 28% amplitude. Centrifugation at 4500 × g for 20 min yielded the supernatant as a sample lysate prepared in HTP mode.
HTP prediction of Vs of tagged esterase/mutant in lysates
96-well microplates were coated with the mcAb in 200 μL coating solution (50 mM sodium carbonate buffer at pH 9.6) by incubation at 37 °C for 30 min and then at 4 °C for 20 h. To correct for nonspecific adsorption, control wells were prepared by treating wells with the coating solution alone. After coating, solutions in wells were discarded and wells were washed three times with 380 μL of the washing buffer (20 mM Tris–HCl buffer at pH 7.4) with continuous vibration of microplates for 2.0 min each time. To block exposed surfaces, wells were further coated with 360 μL solution of BSA at 30 g L−1 for 2 h at 37 °C. Each diluted sample lysate of 200 μL was loaded into wells for 1.0 h binding reaction at 37 °C. Wells were washed with 380 μL of the washing buffer plus 0.05% Tween 20 three times. Finally, 180 μL substrate solution at 0.25 mM was added and microplates were continuously vibrated for 2.0 min; absorbance at 405 nm was then measured with a Biotek ELX 800 microplate reader at 1.5 min intervals and 25 ± 0.5 °C in 30 min. Nonspecific adsorption was corrected with the same diluted sample lysate in control wells. Initial rate was estimated from absorbance below 0.630 and expressed as the total increase of absorbance for 30 min initial rate reaction. The linear range covered an absorbance change of 0.060 to 1.800 in 30 min. The same diluted sample lysate was incubated in a control well to correct potential loss of activity during operation (usually less than 10%).Other analyses and data processing
Protein quantity was measured by the Bradford method.9 The apparent specific activity was the enzyme activity divided by the quantity of total proteins in a sample. Specific activity was the activity divided by the quantity of a purified enzyme. The ratio of the apparent specific activity of a 6His-tagged enzyme in a cell lysate to the approximated specific activity after purification reflected the enzyme abundance. The standard error of estimate was derived from regression analysis. The upper limit of linear response was defined as the largest datum determined with a deviation from the predicted value smaller than three times the standard error of estimate. CV was deduced from standard deviation and mean. Each datum for predicting Vs was the mean from at least five wells. Statistical comparison and regression analysis according to eqn (4) were made with MS Excel 6.0.Acknowledgements
We acknowledge supports from the National Natural Science Foundation of China (no. 81071427), the Natural Science Foundation Project of CQ (no. CSTC2012JJA0057) and the Education Ministry of China (no. 20125503110007).Notes and references
- (a) S. Bershtein and D. S. Tawfik, Curr. Opin. Chem. Biol., 2008, 12, 151 CrossRef CAS PubMed; (b) N. J. Turner, Nat. Chem. Biol., 2009, 5, 567 CrossRef CAS PubMed; (c) C. Jackel and D. Hilvert, Curr. Opin. Biotechnol., 2010, 21, 753 CrossRef PubMed.
- (a) M. Olsen, B. Iverson and G. Georgiou, Curr. Opin. Biotechnol., 2000, 11, 331 CrossRef CAS; (b) N. Cohen, S. Abramov, Y. Dror and A. Freeman, Trends Biotechnol., 2001, 19, 507 CrossRef CAS; (c) P. Soumillion and J. Fastrez, Curr. Opin. Biotechnol., 2001, 12, 387 CrossRef CAS; (d) W. P. Walters and M. Namchuk, Nat. Rev. Drug Discovery, 2003, 2, 259 CrossRef CAS PubMed.
- J. Feng, H. Liu, X. Yang, A. Gao, J. Liao, L. Feng, J. Pu, Y. Xie, G. Long, Y. Li and F. Liao, Chem. Cent. J., 2013, 7, 69 CrossRef PubMed.
- (a) K. Terpe, Appl. Microbiol. Biotechnol., 2003, 60, 523 CrossRef CAS PubMed; (b) H. Block, B. Maertens, A. Spriestersbach, N. Brinker, J. Kubicek, R. Fabis, J. Labahn and F. Schäfer, Methods Enzymol., 2009, 463, 439 CAS.
- (a) F. Liao, W. Liu, Q. Zhou, Z. Zeng and Y. Zuo, Clin. Chim. Acta, 2001, 314, 67 CrossRef CAS; (b) F. Liao, K. Tian, X. Yang, Q. Zhou, Z. Zeng and Y. Zuo, Anal. Bioanal. Chem., 2003, 375, 756 CAS; (c) F. Liao, D. Yang, J. Tang, X. Yang, B. Liu, Y. Zhao, L. Zhao, H. Liao and M. Yu, Clin. Biochem., 2009, 42, 926 CrossRef CAS PubMed.
- (a) H. Liu, X. Yang, L. Liu, J. Dang, Y. Xie, Y. Zhang, J. Pu, G. Long, Y. Li, Y. Yuan, J. Liao and F. Liao, Anal. Chem., 2013, 85, 2143 CrossRef CAS PubMed; (b) H. Liu, X. Yang, J. Dang, L. Liu, X. Hu, J. Pu, G. Long and F. Liao, Anal. Methods, 2013, 5, 5969 RSC.
- Y. J. Choi and C. B. Miguez, Appl. Environ. Microbiol., 2004, 70, 3213 CrossRef CAS PubMed.
- X. Yang, X. Hu, B. Xu, X. Wang, J. Qin, C. He, Y. Xie, Y. Li, L. Liu and F. Liao, Anal. Chem., 2014, 86, 5667–5672 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Including supplementary data. See DOI: 10.1039/c4ra03189j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |