DOI:
10.1039/C4RA03158J
(Paper)
RSC Adv., 2014,
4, 24962-24972
Synthesis of ZnO/Au and ZnO/Ag nanoparticles and their photocatalytic application using UV and visible light†
Received
8th April 2014
, Accepted 20th May 2014
First published on 29th May 2014
Abstract
We report a simple and convenient method for the synthesis of a ZnO/Au and ZnO/Ag heterostructure nanoflower by applying a surfactant mediated route. Initially, pure ZnO nanoflowers have been synthesized followed by Au and Ag deposition on ZnO surface using hydrazine hydrate as reducing agent. Structure, crystallinity, and morphology have been assessed by X-ray diffraction, X-ray photoelectron spectroscopy, and electron microscopy techniques. The influences of the deposited metal nanoparticles (Au and Ag) on the surface of ZnO have been emphasized by applying the as-synthesized nanostructure in dye degradation under illumination of UV and visible light. The basic motivation behind this work is to find a superior photocatalyst, which can work under UV as well as visible light i.e., to cover the whole range of the solar spectrum. Photocatalytic performances of bare ZnO, ZnO/Au, and ZnO/Ag have been studied thoroughly. Photodegradation results under UV and visible light demonstrated that the incorporation of noble metal nanoparticles significantly (or drastically) increases the catalytic efficiency by promoting the photogenerated charge carrier separation. The main advantage of the proposed ZnO/Au and ZnO/Ag semiconductor is that it delays the recombination process of the electron–hole pairs generated by the photon absorption, which in lieu increases the photocatalytic efficiency. It is a challenging issue to fabricate stable photocatalysts which can work under visible light as it covers 43% of sunlight. To investigate the role of photogenerated electrons and holes in dye degradation, scavenging experiments using different scavengers have also been performed.
Introduction
In recent years, scientists have been investigating many semiconductor-assisted photocatalysts due to their ability to degrade a large number of recalcitrant chemicals in aqueous systems with high efficiency. Among those semiconductor assisted photocatalysts, ZnO nanomaterials have extensively been studied due to their high surface area and high photosensitivity.1–4 However, owing to its high band gap (3.37 eV) and large exciton binding energy (60 MeV),1 the use of ZnO in photocatalysis is limited mainly under UV light of wavelength below 385 nm. As the sunlight reaching to the earth's surface contains even less than 5% of it in the form of UV light, the practical application of ZnO photocatalysts is largely restricted. However, modified ZnO with noble metals, such as Pt,5,6 Au,7,8 and Ag9 possess many unique properties such as easy reduction, high chemical stability, and bio affinity. In addition, noble metals also act as an electron sink, which can significantly enhance the photocatalytic activity in both UV and visible light.10–13 Therefore, it is of high practical importance to show how the plasmonic energy can be converted to chemical energy when ZnO nanoflower surface has been functionalized with gold and silver nanoparticles.
Synthesis of zinc oxide nanoparticles have been reported by different groups using various methods such as, hydrothermal synthesis,14,15 alkali precipitation,16,17 thermal decomposition,18 organo-zinc hydrolysis,19 etc. Yang and co-workers reported cetyltrimethylammonium bromide (CTAB) assisted hydrothermal synthesis of ZnO nanoflower using an autoclave at 120 °C.14 Synthesis of ZnO nanorod and prism using zinc foil, NaOH, and CTAB at a temperature ∼160 °C has been reported by Wang and co-workers.20 Low temperature synthesis of various superstructures (ring, sheet, and platelet) of ZnO using CTAB has also been reported by Lee and co-workers.21 Room temperature synthesis of flower-like ZnO nanostructures without using any external capping agent at alkaline pH has been reported by Kumbhakar and co-workers.22 Flower-like morphology of ZnO has been found by Miao and co-workers using lignin amine for 5 h in the 80 °C water bath.23 Hence, from all the reported literatures it has been quite clear that autoclave at elevated temperature as well as room temperature can be a useful method for the synthesis of ZnO particles.24–27 Whereas, within this study, we report a simple approach for the synthesis of ZnO nanoflower at low temperature using CTAB as a growth controlling agent and their surface modification with noble metal nanoparticles, which can work as an active and efficient photocatalyst covering the whole solar spectrum.
Surface modification/decoration of ZnO with noble metal nanoparticles (Au and Ag) offer many potential challenges. Lee and co-workers reported the synthesis of glutathione protected gold nanoparticles on the surface of ZnO and their successful application in phtocatalysis.28 Moreover, they also reported that the size-dependent gold nanoparticles synthesis on ZnO surface and their photocatalytic activity can be controlled by size-dependent gold capacitor.29 Kamat and co-workers reported the synthesis of ZnO/Au nanoparticles and the Fermi level equilibration during the interaction between the semiconductor and noble metal.30 This phenomenon is very much important to detect the role of noble metal in photocatalysis. They have reported the Fermi level shift towards the conduction band in semiconductor in case of ZnO/Au.30 Synthesis of Ag/ZnO heterostructures with different silver content using solvothermal method and their application in photocatalysis has been reported by Zheng and co-workers.31 Worm-like core/shell composites of Ag/ZnO with different silver content under ultrasonic irradiation has been synthesized by Jia and co-workers.32 Silver nanoparticle decoration onto the flower-like morphology of ZnO and their application in photocatalysis has been reported by Jia et al.33 In general, pure ZnO shows less reactivity as photocatalyst due to the lack of electron and hole trapping agents at conduction and valence band, respectively. Upon modification with noble metals (Au and Ag), photocatalytic activity of ZnO significantly increases, as the metal centres acts as an electron sink/trap on semiconductor surface.10–13 Moreover, due to the difference in work function values of different metals, metal semiconductor heterostructure exhibits variable catalytic efficiency in photocatalysis upon irradiation of visible and UV light.9 Therefore, for better understanding of the effect of noble metals on ZnO nanoparticles and their Fermi level shift, we explore a simple and facile approach for the decoration of Au and Ag nanoparticles on ZnO surface and investigate their application in organic pollutant degradation.34,35 However, a comparative study of the decoration of both Au and Ag nanoparticles on ZnO surface and their photocatalytic effect using UV and visible light is still missing which has been reported within this paper.
In this article, we report a solution based one-step synthesis of ZnO nanoflower at relatively lower temperature (∼80 °C), using cetyltrimethylammonium bromide (CTAB) as a growth controlling agent. Afterwards, the noble metals (Au and Ag) were deposited on the surface of ZnO nanoflower using hydrazine hydrate as a reducing agent. The as-synthesized ZnO/Au and ZnO/Ag heterostructure nanoparticles are characterized using UV-vis, PL (photoluminescence), X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), field-emission scanning electron microscope (FESEM), transmission electron microscope (TEM), and energy dispersive spectroscopy (EDS) techniques. The detailed mechanism for the growth of ZnO nanoflower, ZnO/Au, and ZnO/Ag is thoroughly discussed. Photocatalytic performance of the as-prepared catalysts were evaluated by using a cationic dye, methylene blue (MB) and phenol in water, under illumination of UV and visible light. The main importance of this work is to modify ZnO flower surface with gold and silver nanoparticles and to find out their Fermi level shift towards the conduction band of the semiconductor, which can influence the photoactivity of ZnO. The key role of the noble metal (Au and Ag) on ZnO surface is also examined in dye degradation reaction. Finally, scavenging experiment using some scavengers is executed to know the degradation mechanism.
Experimental methods
Chemicals
All chemicals were of analytical grade (AR) and used as purchased without any further purification. Analytical grade chemicals from different suppliers such as, HAuCl4·3H2O (hydrogen tetrachloroaurate trihydrate, Sigma Aldrich), AgNO3 (silver nitrate, ≥99.9%, Sigma Aldrich), NH2·NH2·H2O (hydrazine hydrate, SD fine-chem limited), zinc sulphate (ZnSO4·7H2O, SD fine-chem limited), cetyltrimethylammonium bromide (Spectrochem Pvt. Ltd.), ammonia solution with sp. gr. 0.91 (SD fine-chem limited), and methylene blue (Sigma Aldrich) were used. Phenol, Ammonium oxalate, and tertiary butyl alcohol were purchased from SD fine-chem limited. All solutions were prepared using deionized water.
Synthesis of ZnO nanoparticle
ZnO nanoparticles were synthesized using the following procedure. Firstly, in a 500 ml beaker, equal volume of (4 ml) of zinc sulphate (1 M) and CTAB (1 M) solutions were dissolved in 400 ml water so that the final concentration of both was 0.01 M and stirred well. Afterwards, the whole solution was kept on water bath at ∼80 °C for ∼5 h and without stirring. Secondly, 2 ml of concentrated ammonia solution (directly from the bottle) was added to the above solution and the pH was maintained ∼10. Immediately, the whole solution was cloudy, which was due to the formation of zinc hydroxide. Finally, white powder of ZnO was formed and precipitated. The white precipitate was collected and washed two times with water and dried well in air.
Synthesis of ZnO/Au and ZnO/Ag nanoparticle
Deposition of Au and Ag on ZnO surface was done by the following procedure. At first, in a 100 ml beaker, 100 mg of the as-synthesized white ZnO powder was dispersed well in 10 ml water by sonication for ∼30 min. Secondly, 10 ml of 1 × 10−2 M HAuCl4 was added to the above dispersed ZnO solution, so that the final concentration of HAuCl4 was maintained at 0.5 × 10−2 M with continuous stirring but without addition of any extra growth controlling agent. As a result, in the above solution the weight ratio of ZnO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AuCl4− was 2.5:1. The whole solution was stirred well for ∼2 h so that the HAuCl4 was adsorbed well on ZnO surface. Afterwards, the HAuCl4 adsorbed ZnO precipitate were collected and washed several times with water to drain out the excess HAuCl4 from the solution. In third step, the precipitate was re-dispersed again in 20 ml water followed by addition of 0.2 ml of 1 M hydrazine hydrate within the solution with continuous stirring. Au nanoparticles were deposited on to the surface of ZnO particles which results in a purple coloured precipitate formation. Finally, this purple colour powder was collected and washed with deionised water twice and dried well for further characterization and application.
:AuCl4− was 2.5:1. The whole solution was stirred well for ∼2 h so that the HAuCl4 was adsorbed well on ZnO surface. Afterwards, the HAuCl4 adsorbed ZnO precipitate were collected and washed several times with water to drain out the excess HAuCl4 from the solution. In third step, the precipitate was re-dispersed again in 20 ml water followed by addition of 0.2 ml of 1 M hydrazine hydrate within the solution with continuous stirring. Au nanoparticles were deposited on to the surface of ZnO particles which results in a purple coloured precipitate formation. Finally, this purple colour powder was collected and washed with deionised water twice and dried well for further characterization and application.
ZnO/Ag nanoparticles were synthesized using the same protocol described for ZnO/Au nanoparticles, except AgNO3 was used instead of HAuCl4 where a yellow colour precipitate was obtained.
Photocatalysis study
To examine the photocatalytic activity, MB was chosen as a probe molecule. To study the photocatalytic effect, 4.0 ml of 5 × 10−5 M MB and 50 mg catalyst were collected in 16 ml water. Prior to any irradiation, the aqueous solution of MB and catalyst were agitated continuously at room temperature for ∼40 min using a sonicator, so that MB was adsorbed well on the catalytic surface. After agitation, the reaction mixture was kept under a UV/visible light to initiate the reaction with continuous stirring and the distance between the light source and reaction mixture was fixed at ∼20 cm. The UV light was provided by a UV lamp with major emission at λ = 365 nm whereas visible light was supplied using a tungsten lamp of 60 W, which emits a continuous spectrum of light in between 300–1400 nm. During the photocatalysis investigation, reaction mixture was stirred continuously. Finally, at a regular irradiation time interval, the dye containing solution was quantified by measuring the absorbance using the UV-vis spectroscopy. The as-synthesized nanoparticles were very stable and the morphology was unchanged even after prolonged photodecomposition. To check the reusability, the catalyst particles were collected and washed 3–4 times with deionized water.
Similar photocatalysis study was also performed with phenol using ZnO/Au and ZnO/Ag nanocatalysts. For this study, 0.8 ml of 5 × 10−3 M phenol and 50 mg catalyst were mixed in 20 ml water. After ∼40 min sonication of the reaction mixture, a tungsten lamp of 60 W was used as a visible light source to degrade the phenol.
The efficiency of dye degradation was calculated using the following equation:
| % degradation = (A0 − At)/A0 × 100 |
where,
A0 = initial absorbance and
At = absorbance at time ‘
t’.
Optical characterization
UV-vis absorption spectra were obtained using a Shimadzu UV-1800 spectrophotometer where the cuvette path length was set to 1.0 cm. Aqueous solution was used for background subtraction. Room temperature PL measurement was recorded using a Horiba Jobin Yvon Fluoromax-4 spectrofluorimeter. The excitation and emission slit widths were fixed to 5 nm each and a 325 nm light was used as an excitation wavelength (λex).
Chemical analysis
XPS compositional analysis was carried out using a commercial Omicron EA 125 spectrometer equipped with a seven-channel detection system. As an X-ray source, a monochromatic light of Al-Kα radiation of energy 1486.7 eV has been used. All high resolution spectra were collected with an energy step of 0.1 eV, pass energy of 20 eV and a dwell time of 1 s per step. The emission current was set to 15 mA for all measurements. The XPS chamber base pressure was maintained below <3 × 10−10 mbar during the measurement. Samples were prepared by dropping 10.0 μl aqueous solution of each nanoparticle onto small pieces of conducting carbon tape and dried under dry nitrogen line. To compensate any kind of charging effect, the binding energy of C 1s peak at 284.5 eV has been used as a reference.
Structure and morphology
TEM micrographs were obtained using a JEOL-2010F TEM operating with electron beam of energy of 200 kV. Samples were prepared by placing 3–4 drops of the well dispersed ZnO, ZnO/Au and ZnO/Ag samples on a 300-mesh, carbon-coated Cu grid (EM sciences) and allowing the liquid to evaporate in air. For Au and Ag nanoparticles the particle size distribution was based on 30 randomly selected particles.EDS measurement was carried out with the same instrument and in a particular area of the samples. The phase and the crystallinity of the samples were determined by powder X-ray diffraction (PXRD) using a Rigaku Mini Flex II diffractometer with Cu-Kα radiation at 25 °C with 2θ values ranging from 20–80° and scanning rate of 2° per min. The surface morphology of as-synthesized nanoparticles was characterized using a commercial FESEM instrument from FEI Nova NanoLab combined with focused ion beams. For SEM characterization, sample preparation was similar to that of XPS studies.
Result and discussion
Synthesis and UV-vis analysis of ZnO, ZnO/Au, and ZnO/Ag nanoparticles
The syntheses of ZnO/Au and ZnO/Ag have been described previously and are reviewed in the Experimental section. Briefly, ZnO nanoparticles are first synthesized using CTAB surfactant as a growth controlling agent. Throughout the synthesis, the pH of the solution is adjusted to ∼10 with ammonia solution. Afterwards, gold and silver nanoparticles are deposited on the surface of ZnO using hydrazine hydrate as a reducing agent. All as-synthesized nanoparticles are very stable and used for other characterizations which have been discussed in the following.
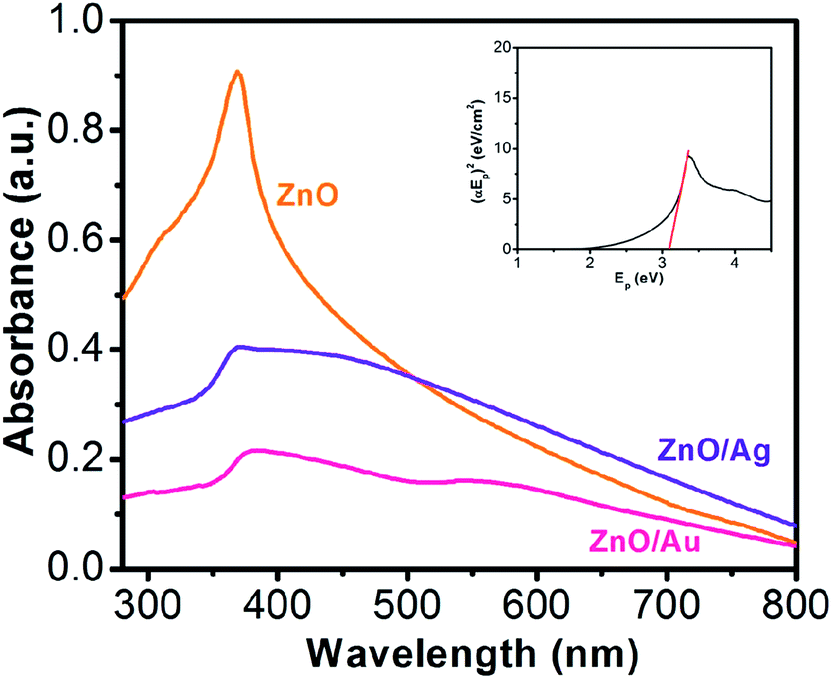
Fig. 1 shows the absorption spectroscopy of the ZnO, ZnO/Au, and ZnO/Ag nanoparticles in the ultraviolet-visible spectral region. ZnO exhibits a sharp band at 369 nm, which corresponds to the formation of ZnO particles. From the absorption spectrum of ZnO an estimated optical band gap can be derived using the following equation:
where,
α stands for the absorption coefficient,
K is a constant,
Ep is the discrete photo energy, and
Eg is the band gap energy. A classical Tauc approach is further employed to estimate the
Eg value of ZnO nanoparticles.
36–38 A plot of (
αEp)
2 vs. Ep is shown within the inset of
Fig. 1. The extrapolated straight line of this plot meets the
Ep axis at
α = 0 which represents the absorption edge energy corresponds to the band gap (
Eg) of the material. The band gap value of ZnO from the experimental data was calculated to be 3.12 eV, which is in good agreement with the value reported in the literature.
39 In case of ZnO/Au, spectroscopy shows two bands at 378 nm and 560 nm, respectively. The band position at 378 nm corresponds to the presence of ZnO whereas the other band represents the formation of Au nanoparticles. For ZnO/Ag samples a broad band at 450 nm, representative of the Ag nanoparticles appears along with the unshifted band at 369 nm for ZnO particles. The shift in ZnO band position in the case of ZnO/Au nanoparticles can be attributed to the higher electronegativity of gold compare to that of silver. As a result, gold withdraws electron density more towards itself, which finally affect the movement of the band position of ZnO. In addition, a significant decrease in absorbance intensity of ZnO peak at 369 nm for ZnO/Au and ZnO/Ag have also been observed, which indicates the presence of gold and silver on ZnO surface. Therefore, the presence of UV and visible absorbance maxima both for ZnO/Au and ZnO/Ag clearly demonstrates that the material becomes photoactive in both UV and visible light region.
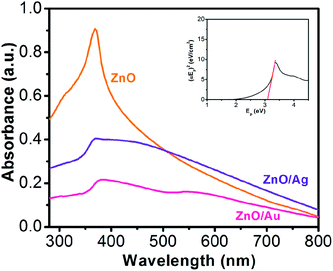 |
| | Fig. 1 UV-Vis spectra for ZnO, ZnO/Au, and ZnO/Ag nanoparticles, where all the powder samples were dispersed in water and sonicated for ∼20 min. Aqueous solution was used for background subtraction whereas. 1.0 cm cuvette was used for UV-Vis measurement. Here, all absorbance values are in arbitrary unit and each spectrum corresponds to different absorbance (not in relative scale). From the UV-vis spectrum of ZnO, inset shows (αEp)2 vs. Ep plot for band gap (Eg) calculation of ZnO nanoparticles. | |
Photoluminescence study of ZnO, ZnO/Au, and ZnO/Ag nanoparticles
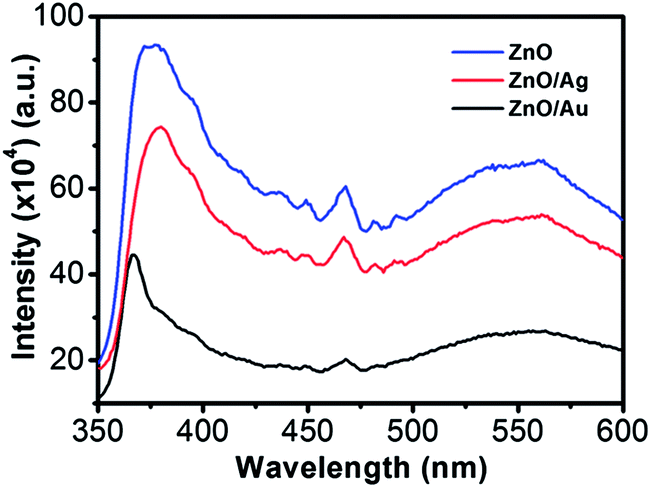
Room-temperature PL spectra of the ZnO, ZnO/Au, and ZnO/Ag are shown in Fig. 2, which was obtained with an excitation wavelength of 325 nm. For all the nanoparticles three bands are observed in PL spectrum: (a) UV near-band-edge emission peak at 378 nm, (b) small band at 467 nm, and (c) a broad green light emission at 560 nm. The green emission peak is commonly referred to a deep level of trap-state emission.40 More precisely, the green transition has been attributed to the singly ionized oxygen vacancy in ZnO and the emission results from the radiative recombination of a photogenerated hole with an electron occupying the oxygen vacancy.40,41 The stronger intensity suggests the presence of higher singly ionized oxygen vacancies in ZnO.29 When ZnO is modified with metal nanoparticles, the intensity of all the three bands decreases, which states that the electrons and holes are more separated and recombination process is inhibited. The relatively lower intensity bands for the case of ZnO/Au proves the inhibited electron–hole pair recombination is maximum for the ZnO/Au nanoparticles. However, the shifting of UV near-band-edge emission peak at 367 nm for ZnO/Au is still not well understood.
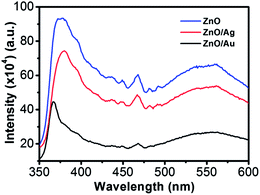 |
| | Fig. 2 Room temperature photoluminescence spectra of ZnO, ZnO/Au, and ZnO/Ag nanoparticles. All the powder samples were well dispersed in water using a sonicator for PL measurements using an excitation wavelength (λex) of 325 nm. | |
XRD analysis of ZnO, ZnO/Au, and ZnO/Ag nanoparticles
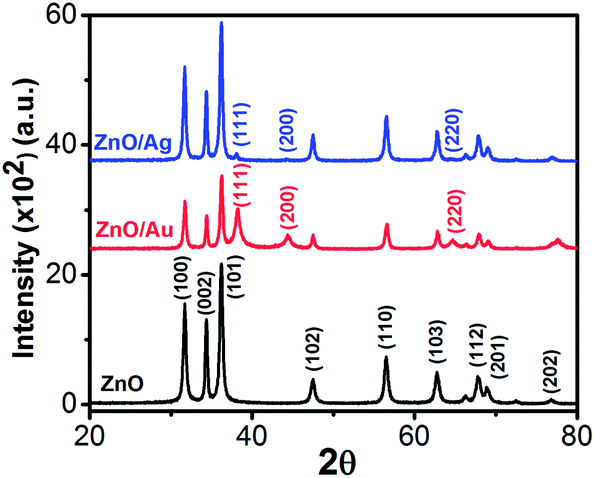
The crystallinity, phase, and purity of the as-prepared samples were determined by powder X-ray diffraction (PXRD). Typical PXRD patterns of the as-synthesized ZnO, ZnO/Au, and ZnO/Ag particles are shown in Fig. 3. Nine major reflection peaks are appeared at 2θ values of 31.80°, 34.41°, 36.21°, 47.52°, 56.53°, 62.74°, 67.80°, 69.11°, and 77.46° which corresponds to the (100), (002), (101), (102), (110), (103), (112), (201), and (202) crystal planes of the hexagonal ZnO structure, respectively. All these peaks of the ZnO nanoparticles can be indexed to wurtzite (hexagonal) structure of ZnO (JCPDS card no. 36-1451).14 Absence of characteristic impurity peaks such as Zn(OH)2 and unhydrolyzed Zn(II)-sulphate indicate a high quality ZnO nanoparticles.
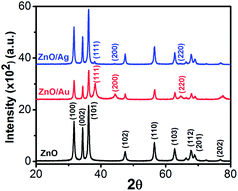 |
| | Fig. 3 Powder X-ray diffraction (PXRD) patterns of ZnO, ZnO/Au, and ZnO/Ag nanoparticles. During scan 2θ values varies from 20–80° and the scanning rate was fixed at 2° per min. | |
The PXRD patterns of ZnO/Au exhibits three additional peaks at 2θ values of 38.23°, 44.38°, and 64.7° corresponds to the (111), (200), and (220) crystal planes of Au, respectively, which confirm the formation of gold nanoparticles on ZnO surface.42 PXRD pattern of ZnO/Ag shows a clear peak at 38.05°, which is due to the presence of (111) crystal plane of silver particle on ZnO surface. The other two peaks at 44.38° and 64.7° for (200) and (220) crystal planes of Ag are not clear.31 All these PXRD findings clearly indicate the formation of metal nanoparticles on ZnO surface. Au and Ag particle sizes were calculated using Debye–Scherrer formula, which represents 20 nm and 32 nm for Au and Ag nanoparticles, respectively.
SEM, TEM, and EDS analysis of ZnO, ZnO/Au, and ZnO/Ag nanoparticles
The morphological analysis of the as-synthesized nanoparticle samples were carried out using an FESEM, which is depicted in Fig. 4. ZnO particles show flower shaped structures having diameter of 600 nm (length) of the petal. Fig. 4a and b show a surface overview and a closer look FESEM images of ZnO particles, respectively, which contain a sharp petal with breadth of 220 nm for only one petal in ZnO nanoflower. High resolution SEM image clearly shows a little surface roughening of the ZnO flower petals. A similar kind of flower-like structure has also been reported by Yang and co-workers, where they used a saturated CTAB aqueous solution at ∼120 °C for 20 h and a Teflon-lined stainless steel autoclave.14 Whereas, in this study, ZnO particles were synthesized in a much simpler way, using water bath at ∼80 °C for 5 h.
 |
| | Fig. 4 FESEM images of ZnO nanoparticles: (a) large scale overview and (b) closer look of bare ZnO nanoflowers. High-resolution FESEM images of (c) ZnO/Au and (d) ZnO/Ag. Particle sizes for Au and Ag were measured using randomly chosen ∼30 particles. | |
FESEM images of gold and silver nanoparticles, deposited on ZnO surfaces are shown in Fig. 4c and d, respectively. Randomly scattered spherical particles with uniform size-distribution of gold and silver were nicely resolved on ZnO surface. From the FESEM images, it has been observed that the average particle diameter for gold is about 30 nm, whereas for silver it rises to 40 nm. Hence, from the above findings we can conclude that the synthesis of ZnO/Au and ZnO/Ag nanoparticles by a simple wet-chemical method, using hydrazine hydrate as reducing agent, is very much useful.
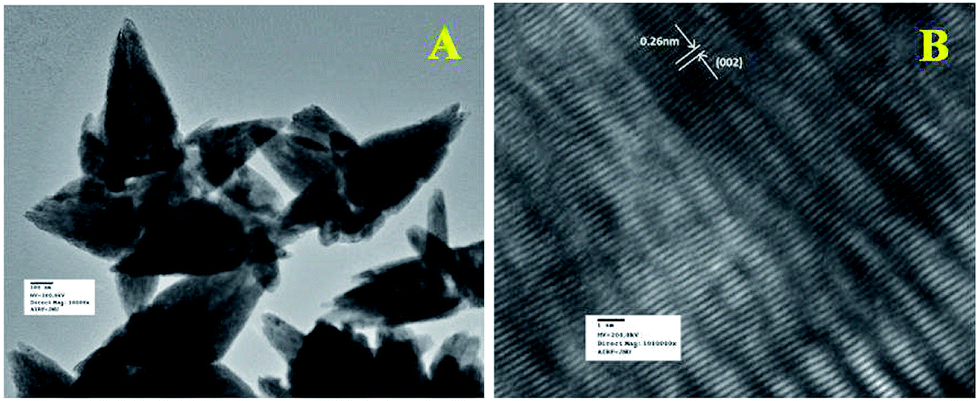
Fig. 5a shows the TEM image of flower shape structure of the ZnO nanoparticles. TEM result also confirms the presence of sharp petal with an average size of 600 nm long (length) and 230 nm (breadth) diameter as well as little surface roughening of the petal structure. High resolution TEM image of ZnO particle is shown in Fig. 5b, which clearly depicts lattice fringes of ZnO with an interplaner spacing of ‘d’ = 0.26 nm, which matches well with the spacing between (002) crystal planes of ZnO. This finding of (002) plane oriented growth of ZnO corroborates well with the earlier PXRD results.
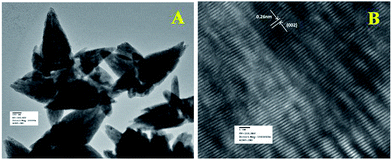 |
| | Fig. 5 (a) TEM and (b) HRTEM images of ZnO nanoparticles. HRTEM image was used to calculate the fringe spacing. | |
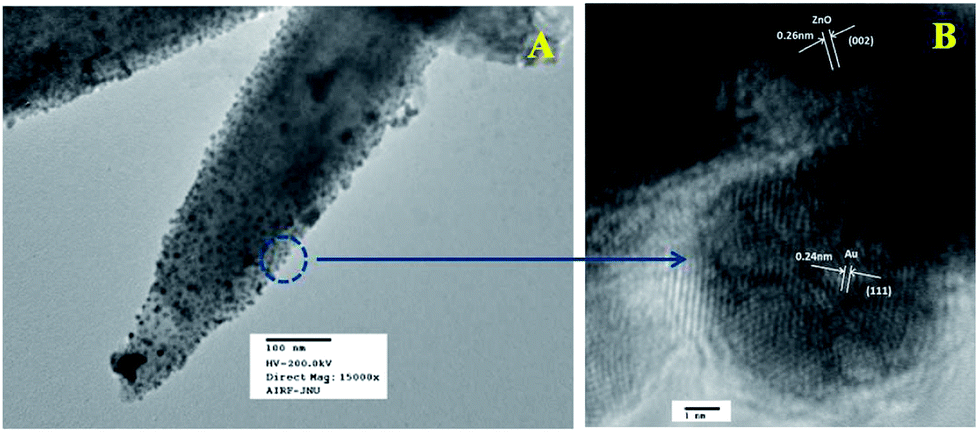
Fig. 6a represents the TEM image of ZnO/Au nanoparticles, which strongly suggests that the formation of spherical gold nanoparticles randomly distributed on the surface of ZnO. An HRTEM image of ZnO/Au is shown in Fig. 6b, which further confirms the presence of both ZnO and gold nanoparticles. The interplaner spacing ‘d’ of 0.24 nm and 0.26 nm exhibits the presence of (111) plane of gold nanoparticles on (002) plane of ZnO surface, respectively. A similar kind of TEM and HRTEM images of ZnO/Ag are presented in Fig. 7a and b. It is observed that, the spherical silver particles are well distributed on ZnO surface. A clear contrast of fringe spacing was observed between the ZnO and silver particles. Separate lattice fringes were observed for ZnO and silver with the value of ‘d’ of 0.26 nm and 0.23 nm, which can be assigned to the presence (111) planes of silver on (002) ZnO surface. All TEM images for the as-synthesized particles are in good agreement with FESEM findings. Both FESEM and TEM images clearly indicate a higher density of metal nanoparticles for of ZnO/Au compare to the ZnO/Ag.
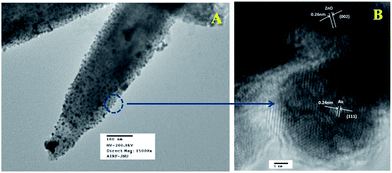 |
| | Fig. 6 (a) TEM and (b) HRTEM images of ZnO/Au nanoparticles. Fringe spacing for ZnO and Au were separately calculated using HRTEM image. | |
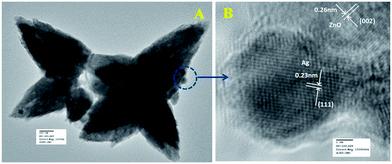 |
| | Fig. 7 (a) TEM and (b) HRTEM images of ZnO/Ag nanoparticles. Fringe spacing for ZnO and Ag were separately calculated using HRTEM image. | |
Finally, EDS analysis of ZnO/Au and ZnO/Ag for selected area also confirms the presence of Zn, O, Au, and Ag, which is shown in ESI (Fig. S1†). The EDS signal of Au and Ag are very uniform in the observed area, indicating the completely random distribution of gold and silver nanoparticle on ZnO surface.
XPS analysis of ZnO, ZnO/Au, and ZnO/Ag nanoparticles
As a surface monitoring technique, XPS was employed to further support the formation of ZnO, ZnO/Au, and ZnO/Ag nanoparticles. The high-resolution XPS spectra of Zn2+ and O2− regions of ZnO particles, synthesized by wet chemical technique are shown in ESI (Fig. S2a and b†). The binding energy positions for Zn 2p3/2 and Zn 2p1/2 are obtained at 1022.1 eV and 1045.1 eV, respectively. The difference in binding energy between two Zn peaks is about 23.0, which matches well with the earlier reported value.42 The O1s peak shows the binding energy position at 530.6 eV. All these findings clearly suggest the formation of ZnO.42,43
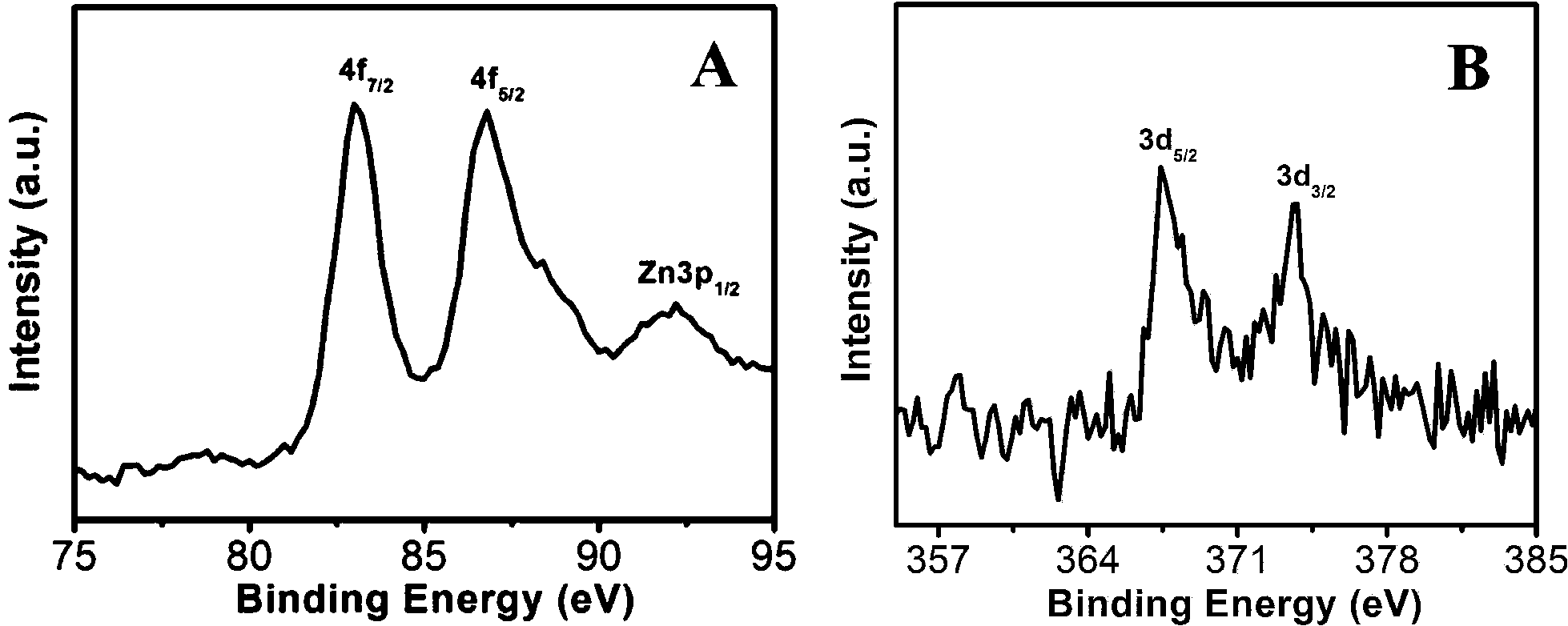
The high-resolution XPS spectra of ZnO/Au and ZnO/Ag are shown in Fig. 8a and b. The Au 4f7/2 and 4f5/2 binding energy peaks are centred around 83.04 eV and 87.14 eV, respectively, which are consistent with those for bulk Au at 83.95 eV and 87.68 eV, indicating the formation of Au(0) nanoparticle on the surface of ZnO using hydrazine hydrate as reducing agent.44,45 Similarly, for ZnO/Ag, the binding energy peaks are located at 373.71 eV (3d3/2) and 368.33 eV (3d5/2), favouring the formation of Ag(0) nanoparticles. The binding energies for Ag(0) are matches well with the reported literature values for bulk Ag at 373.4 eV and 368.22 eV, respectively.44,46 From these binding energy values of gold and silver it has been observed that, there is negligible shift of binding energies (gold and silver) on ZnO surface as compared to their bulk.
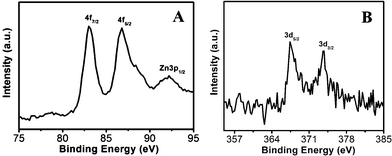 |
| | Fig. 8 High-resolution XPS spectra of (a) ZnO/Au and (b) ZnO/Ag samples deposited on conducting carbon tapes. XPS peak positions were referenced to the C 1s peak at 284.5 eV. | |
Mechanism of formation of ZnO, ZnO/Au, and ZnO/Ag nanoparticles
The mechanism of the formation of ZnO particles in alkaline medium is given below. At alkaline pH, available OH− ion in the aqueous solution initially reacts with Zn2+ ion and introduces the formation of Zn(OH)2. At pH 10, Zn(OH)2 is the main colloid species in the solution. During hydrolysis at ∼80 °C, part of Zn(OH)2 colloid dissolves into Zn2+ and OH− within the solution, which creates the formation of ZnO nuclei. Furthermore, at alkaline pH, Zn(OH)2 further reacts with OH− ion and therefore [Zn(OH)4]2− is formed. Finally, at elevated reaction temperature (∼80 °C), the growth of ZnO nanoflower initiates in presence of CTAB due to the coulomb attraction between the cationic charge CTAB and anionic [Zn(OH)4]2− species.14 The overall reaction for the formation of ZnO is shown below.
| Zn(OH)2 + 2OH− → [Zn(OH)4]2− |
| [Zn(OH)4]2− → ZnO + OH− + H2O |
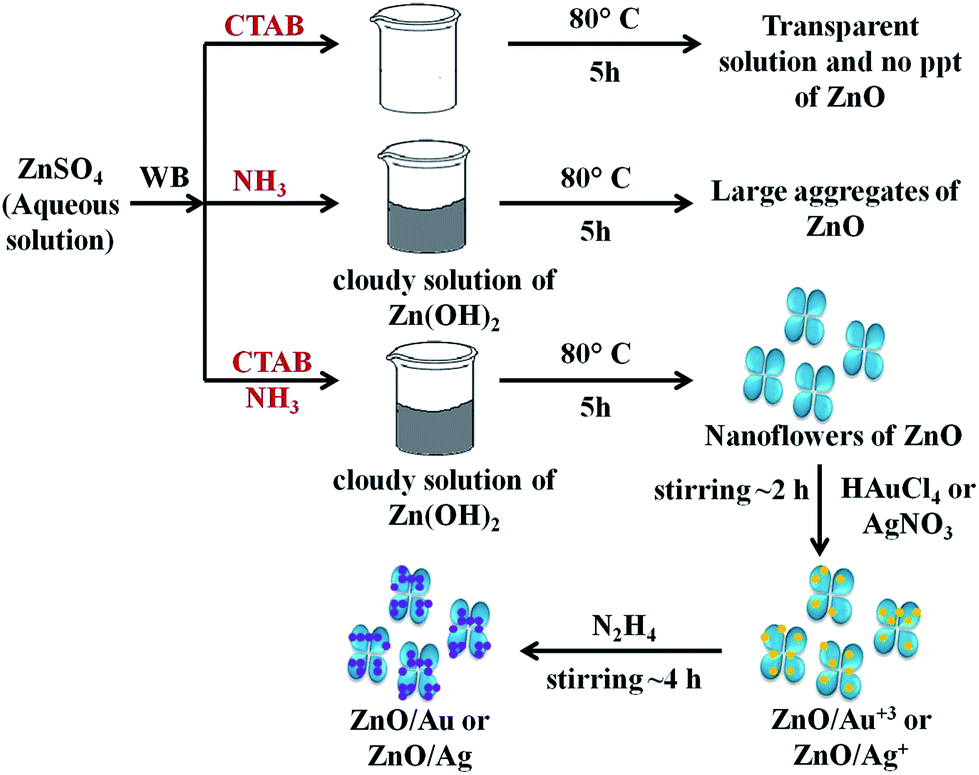
To understand the effect of alkaline medium on formation of ZnO particle, a similar kind of reaction was also carried out in absence of ammonia solution. Surprisingly, no ZnO particle was found (Scheme 1). In case of surfactant assisted synthesis of ZnO, surfactant plays a crucial role and acts as a driving force for the formation of ZnO nanoflower.14 Therefore, to know the role of CTAB in nanoflower formation, hydrothermal experiment was conducted without CTAB, which results in the formation of largely agglomerated ZnO particles instead of a flower-like morphology. The FESEM image of the agglomerated form of ZnO is shown in Fig. S3.† Without the CTAB, there was no growth directing agent. As a result, larger particles are randomly deposited. From Fig. S3,† it is well understood that the role of CTAB is very important for the formation of ZnO flower. Therefore, CTAB is very much essential to generate the flower like morphology of ZnO. Yang and co-workers also reported the formation of pointed sword-like ZnO nanoparticle (∼100 nm) instead of a flower-like morphology, without using the CTAB.14 The same group also reported at lower growth temperature (120 °C), there was not enough driving force for the growth of similar sword-like ZnO nanorods.14
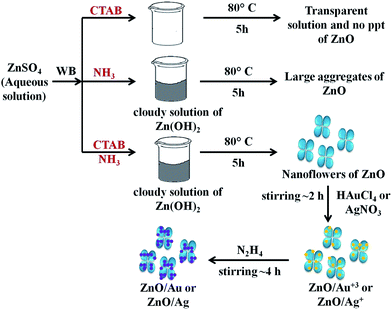 |
| | Scheme 1 The overall mechanism of ZnO/Au and ZnO/Ag nanoparticle formation on ZnO nanoflower surface. Here, WB represents water bath. | |
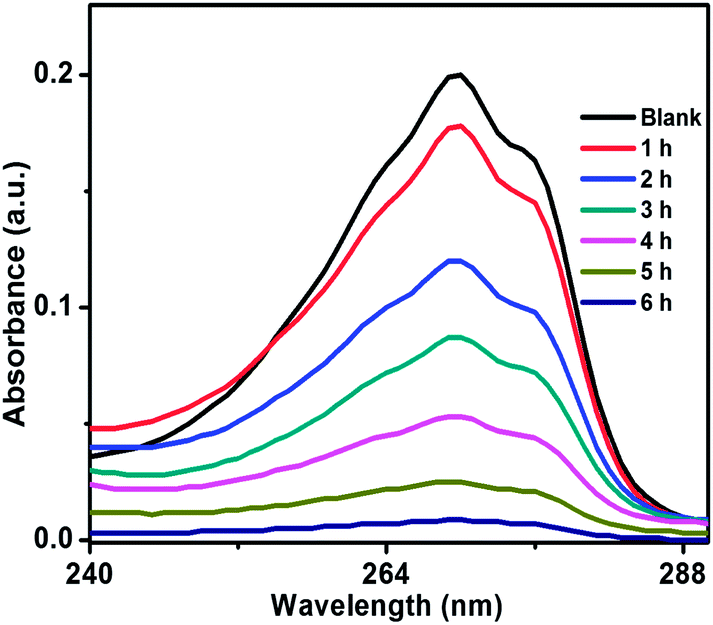
In order to comprehend the complete growth mechanism of ZnO nanostructures in presence of CTAB, a time-dependent analysis of growth features was conducted using FESEM analysis as can be seen in Fig. 9. During synthesis, ZnO powder samples were collected at intervals of 1 h, 3 h, and 5 h, respectively, from the reaction container. Fig. 9a represents the FESEM image of ZnO after 1 h of synthesis, which shows the formation of spherical particles and initiates the formation of ZnO flower. After 3 h of synthesis, ZnO nanoflower formation was started but the growth uniformity was not observed (Fig. 9b). Finally, the complete growth/formation of ZnO flower was ended after 5 h of synthesis, as shown in Fig. 9c. Even longer synthesis does not make any significant changes of the ZnO flower morphology. Therefore, the optimization of reaction time and the role of CTAB during the formation of ZnO were confirmed by growth study.
 |
| | Fig. 9 FESEM images of ZnO nanostructure synthesis after (a) 1 h, (b) 3 h, and (c) 5 h, respectively. | |
For the formation of ZnO/Au and ZnO/Ag particles, the as-synthesized ZnO nanoparticles were well dispersed in aqueous medium and as source materials of Au and Ag nanoparticle, HAuCl4 and AgNO3 were used. Weak van der Waals force or electrostatic interaction may act as a driving force behind the absorption of metal ions on the surface of ZnO. Finally, the adsorbed AuCl4− and Ag+ ions were reduced and deposited randomly on the surface of ZnO using hydrazine hydrate as a reducing agent. Though similar synthetic approaches were used for Au and Ag, the density of deposited Au nanoparticles was higher than that of Ag, which presumably due to higher affinity of AuCl4− than Ag+ towards ZnO surface. This higher affinity of AuCl4− ion could be explained from zeta-potential analysis of ZnO particles. The zeta-potential value of ZnO is (+)25.4 ± 0.63 mV, which confirms a positively charged ZnO surface which is stabilized in the ambient of cationic charge CTAB.14 Hence, a strong electrostatic interaction between the positively charged ZnO and negatively charged AuCl4− is expected which results in a high density of Au nanoparticle on ZnO surface. Whereas, on the other hand, Coulomb repulsion between Ag+ ion and ZnO significantly exists. However, the diffusion energy successfully overcomes the electrostatic repulsion and finally Ag nanoparticles can deposit on ZnO surface. It is quite clear that the driving force for Ag deposition is much weaker compare to the Au, and hence a lower particle density for Ag is observed. Zhu and co-workers also reported a high density of Au nanoparticles on ZnO using electrochemical deposition.42 The overall mechanism behind the formation of ZnO/Au and ZnO/Ag nanoparticle on ZnO nanoflower are explained as can be find in Scheme 1.
To substantially understand the role of hydrazine hydrate, a similar kind of experiment was carried with sodium borohydride as reducing agent. A large and agglomerated particles of Au and Ag were observed on the surface of ZnO nanoflower. Fig. S4† shows the FESEM image of ZnO/Au using sodium borohydride where the Au nanoparticle appear in large, non-uniform, and aggregated nature. Trisodium citrate and ascorbic acid were also used as reducing agent but there were no desirable nanoparticle formation in the followed reaction condition. Therefore, it is well understood from our findings that hydrazine hydrate can be used as a reducing agent to deposit uniform Au and Ag nanoparticles on ZnO surface.
Photocatalytic activity of ZnO, ZnO/Au, and ZnO/Ag nanoparticles
In order to explore the effectiveness of as-synthesized nanoparticles on dye degradation using UV and visible light, a cationic dye, methylene blue (MB) (a standard model water contaminant) were used for this study. Photocatalytic activity of nanoparticles not only correlated with electron transportation but also depends on other factors like surface area, size, radiation sources, etc. ZnO, having a high band gap energy, is not a visible light catalyst. Therefore, decoration of ZnO surface with metal nanoparticle (Au and Ag) is shown to be a very useful technique to make it a visible light active material. Tungsten lamp of 60 W, which emit a continuous spectrum of light in between 300–1400 nm was used as a visible light source.
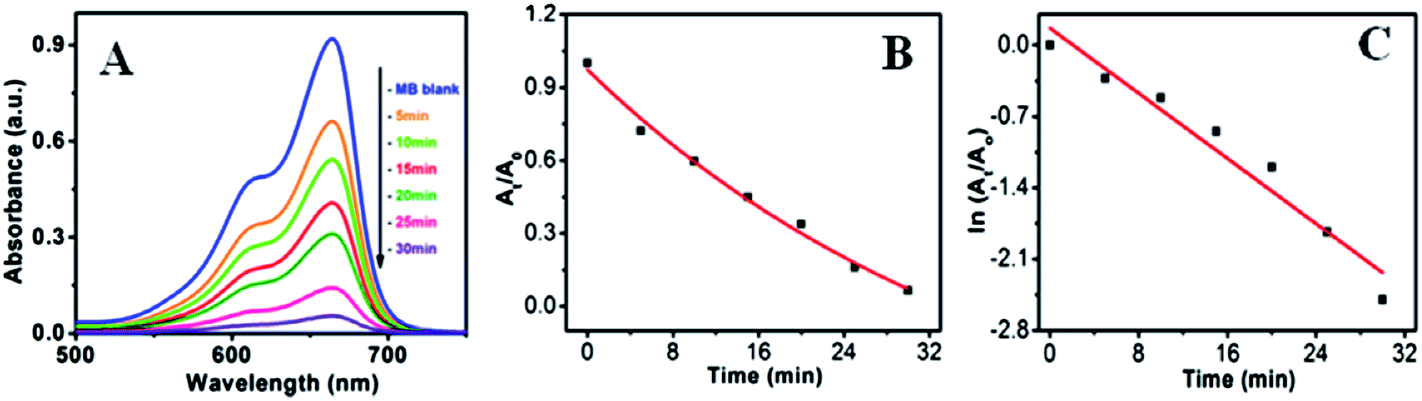
Fig. 10a–c shows the visible light active dye degradation results using 50 mg ZnO/Au as a catalyst where tungsten lamp (60 W) was used as a visible light source. Under the experimental condition, ZnO/Au exhibits 99% dye decomposition within 30 min, with a rate constant of 0.8 × 10−1 min−1. It is known that the concentration of the dye at different time is proportional to the absorbance of the dye solution and therefore, exponential nature of absorbance vs. time (At/A0 vs. t) plot confirms the pseudo first order kinetics of the reaction, as shown in Fig. 10b. From the logarithmic plot of absorbance vs. time (lnAt/A0 vs. t) the degradation rate constant ‘k’ was calculated, which is found to be 0.8 × 10−1 min−1, significantly higher than the earlier reported values for MB dye decomposition (Fig. 10c). It can be noticed that negligible amount of dye degradation occurs in visible light using bare ZnO particle. Even, without any catalyst MB itself has less absorption (∼9% only) in visible light. Whereas, using ZnO/Ag catalyst, 40% of dye degradation can be achieved within 2 h under same visible light, with a rate constant of 0.04 × 10−1 min−1 (Fig. S5a and b†). Afterwards, there is no change in dye degradation using ZnO/Ag and in visible light medium. Similar dye degradation reaction was also carried out in presence of ZnO, ZnO/Au, and ZnO/Ag catalysts in dark condition, which shows there is no change in the intensity of MB even after 3 h (Fig. S6†). This experiment confirms that the dye is not getting adsorbed on the surface of catalyst. Therefore, we can conclude that the incorporation of Au nanoparticles on ZnO surface can drastically enhanced the dye degradation rate up to 20 times as compared to Ag nanoparticle incorporation using visible light. The higher degradation rate for metal induced ZnO can be explained in terms of better separation of the electrons and holes and higher inhibition towards their recombination. This phenomenon of ZnO/Au is in accordance with the PL result, which proves the higher inhibition of electron–hole pair recombination for ZnO/Au nanoparticles. From all these results it is well understood that the modified ZnO with Au and Ag metal nanoparticles shows significantly higher photocatalytic efficiency in visible light region.
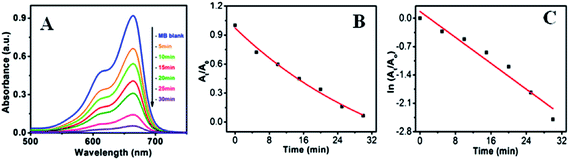 |
| | Fig. 10 UV-vis spectra of (a) degradation of MB dye in presence of ZnO/Au under visible light (b) At/A0 vs. time (min) plot and (c) lnAt/A0 vs. time (min) plot. During the photocatalysis measurements, reaction mixture was stirred continuously. Conditions: [MB] = 10−5 M and amount of catalyst = 50 mg. | |
Photocatalytic activity of ZnO/Au and ZnO/Ag was also performed using another organic pollutant, phenol, under a visible light irradiation. Phenol, a colourless pollutant, was chosen as it has no absorption in the visible light range. Higher concentration of phenol, 2 × 10−4 M and 50 mg catalyst were used for this degradation study. Fig. 11 and S7† exhibits the decomposition of phenol with time, using ZnO/Au and ZnO/Ag catalyst, respectively. As shown in Fig. 11, ∼96% degradation of phenol can be achieved after 6 h using ZnO/Au with a rate constant of 0.51 min−1. Whereas, ∼72% of degradation under visible light is found with ZnO/Ag as shown in Fig. S7.† Using ZnO/Ag, the rate constant value was 0.20 min−1. Therefore, a significantly faster photocatalytic performance was observed with ZnO/Au, 2.5 times higher than ZnO/Ag. The above result proves that under visible light ZnO/Au can be a better catalyst than ZnO/Ag, which is matching well with the earlier MB degradation result.
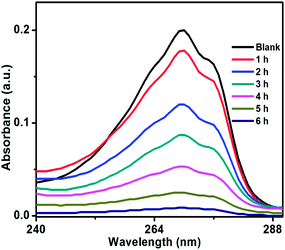 |
| | Fig. 11 UV-vis spectrum of decomposition of phenol using ZnO/Au catalyst, under irradiation of visible light. Conditions: [phenol] = 2 × 10−4 M and amount of catalyst = 50 mg. | |
In case of dye degradation using pure ZnO nanoparticles, a relatively higher photon energy (UV-light of wavelength 365 nm) as well as much longer exposure time is required for ∼50% of decomposition. Whereas, under illumination of UV-light for 30 min, ZnO/Au can successfully degrade up to 99% of the dye. In the case of ZnO/Ag nanoparticles, 95% degradation can be achieved for exposure duration of 3 h. UV-vis spectra of MB dye degradation with ZnO, ZnO/Au, and ZnO/Ag using UV light are compared within the ESI (Fig. S8–S10†). From the kinetic plots of dye degradation with ZnO particles (At/A0 vs. t and lnAt/A0 vs. t) it is quite clear that the reaction follows pseudo first order and the rate constant value is found to be 0.04 × 10−1 min−1. Using ZnO/Au, the dye degradation rate constant value is drastically improved to 0.9 × 10−1 min−1 while following the similar pseudo first order kinetics. A similar kinetics and rate constant value (0.15 × 10−1 min−1) is also observed with ZnO/Ag particles, which can be seen in Fig. S10a and b.† A comparative study of lnAt/A0 vs. time plot for the as-synthesized particles for MB dye decomposition using UV light is shown in Fig. S11a and b.† This result shows that Au decorated ZnO provide maximum rate constants compare to that of ZnO/Ag and bare ZnO, which is in good agreement with the reported literature.10,42
Photocatalytic mechanism
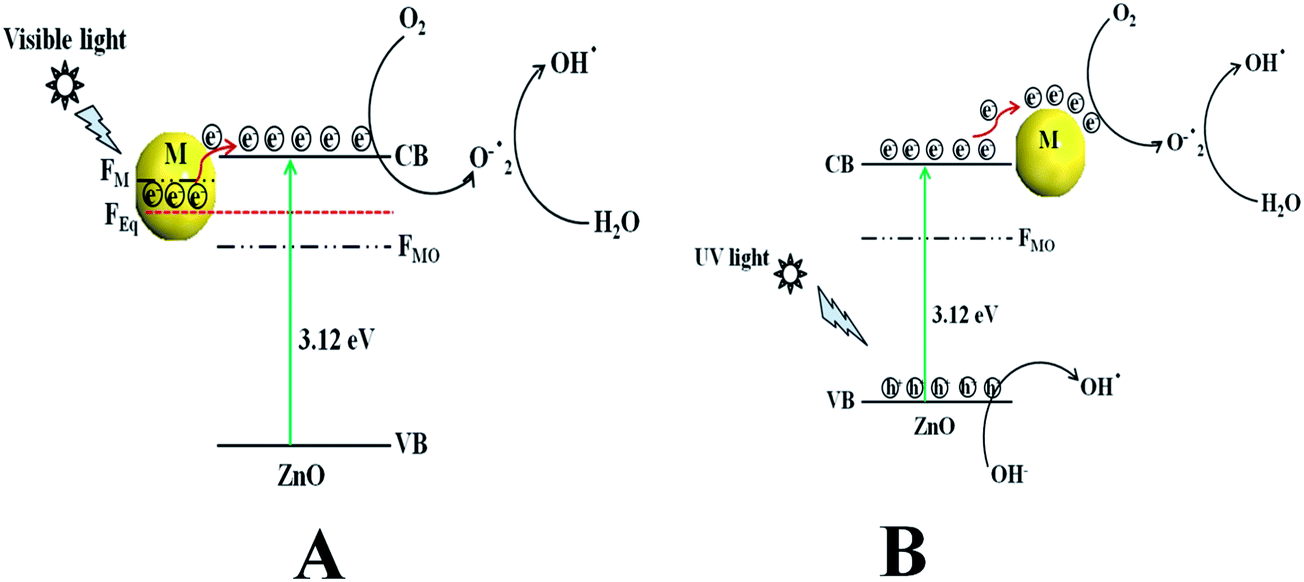
Scheme 2a represents the overall photocatalytic degradation mechanism under irradiation with visible light. The as-obtained visible light photocatalysis result confirms that ZnO/Au is a better photocatalyst than ZnO/Ag and ZnO particles, which can be explained on the basis of work function. The work function values of ZnO, Au, and Ag are reported as 5.2 eV, 5.0 eV, and 4.26 eV, respectively.28,47–50 Normally, a Schottky barrier will form, when two materials with different work functions are combined. Therefore, electrons will transfer from the lower work function material to the higher work function material.30,47–50 In case of Au and Ag the Fermi level (FM) is higher than ZnO Fermi level (FMO) because of its higher work function. This directs the transfer of electron from the Au and Ag Fermi level to the ZnO Fermi level, till it reaches the equilibrium and form a new equilibrium Fermi level (FEq). Upon irradiation with visible light, the electrons in FEq were injected quickly to the conduction band (CB) of ZnO via a surface phonon resonance (SPR) mechanism, leaving behind holes on the metal surface. Now, the injected electrons in CB reacts with dissolve oxygen to produce superoxide radical anion (O2˙−), which can subsequently react with H2O to give hydroxyl radicals (OH˙) in a consecutive reaction pathway.31,49 These superoxide radical anion and hydroxyl radicals are responsible for the decomposition of MB dye and phenol pollutant under visible light irradiation.28 Therefore, due to higher work function of Au as compared to Ag, ZnO/Au can act as a better photocatalyst than ZnO/Ag.47–49 As the formation of reactive radical species is much faster in ZnO/Au compared to the ZnO/Ag, which is also directly correlated with photocatalytic activity, the ZnO/Au particle shows a superior activity. This result is well corroborated with the lower intensity of ZnO/Au than ZnO/Ag in PL study (Fig. 2).
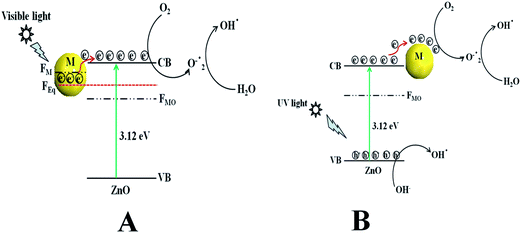 |
| | Scheme 2 The overall dye degradation mechanism under irradiation of (a) visible light and (b) UV light, with ZnO/Au and ZnO/Ag nanoparticles. Here, VB = valence band, CB = conduction band, FMO = Fermi level of metal oxide, FM = Fermi level of metal, FEq = Fermi level of equilibrium, and M = Au and Ag. | |
Scheme 2b demonstrates the degradation mechanism upon irradiation with UV light using ZnO, ZnO/Au, and ZnO/Ag catalysts. In case of ZnO, during illumination with UV light valence band (VB) electrons get excited and move to the CB, leaving behind holes (h+) in the VB of the semiconductor particles. Then the excited electrons in CB react with dissolve oxygen to produce superoxide radical anions (O2˙−), which can subsequently react with H2O to provide OH˙ radicals in a consecutive reaction pathway.31 At the same time, the holes in VB react with surface OH− ion or adsorbed H2O molecule to produce the reactive OH˙ radicals. These O2˙− and OH˙ radicals are also responsible for the further degradation of dye molecules.9,51 As a result, the dye molecules finally degrade to CO2 and H2O. On the other hand, with ZnO/Au and ZnO/Ag, the presence of Au and Ag nanoparticles effectively adsorb electrons from CB and hence prevent the immediate recombination process.52 Here, Au and Ag nanoparticles are used as surface traps, which usually capture the electrons from the ZnO surface and utilises for dye degradation.10–13 Successive electron transfer will be favourable with the suitable junction formation between ZnO and metal nanoparticle (Fig. 6b and 7b). Therefore, the deposited metal nanoparticles (Au and Ag) can act as an electron sink or trap on the surface of ZnO particles.30 Under the same conditions, the higher work function value of Au as compared to Ag make ZnO/Au more-effective for electron acceptor.47–49 As a result, ZnO/Au exhibits a better photocatalytic efficiency in comparison with ZnO/Ag and ZnO. This result also in line with PL study of ZnO/Au and ZnO/Ag particles (Fig. 2). The overall reaction during photocatalysis with visible and UV light are shown below.
| ZnO + hv(UV) → ZnO(eCB− + hVB+) |
| ZnO/M + hv(visible) → ZnO/M*(e−) |
| ZnO/M*(e−) → ZnO(eCB−) + M |
| ˙OH + MB/phenol → decomposed products |
To further investigate the photocatalytic mechanism with better clarity or to understand the actual involvement of the species (e− or h+) for the dye degradation, several scavenging experiments were systematically performed. Ammonium oxalate (AO) and tertiary butyl alcohol (TBA) were used for hole and electron scavenger, respectively. While doing the photocatalysis reaction, 0.1 g of AO and 2 ml of TBA were added to the solution, prior to any addition of catalyst while keeping the other parameters unaltered. It can be noted that the photocatalytic activity of ZnO/Au decreases significantly with the addition of both AO and TBA under a visible light source. A significant decrease is also noticed when AO was added, which behave as hole scavenger. For ZnO/Au, all photocatalytic studies were performed for 30 min. In presence of AO, ZnO/Au catalyst shows 36.67% dye degradation when illuminated with visible light, whereas, with TBA 86.82% dye degradation was observed. Therefore, from this finding it can be concluded that the holes play an important role towards dye degradation under visible light. On the other hand, in presence of UV-light, photodegradation rate is significantly decreased for both ZnO/Au and ZnO/Ag when AO and TBA were added. In presence of AO and TBA degradation of only 39.41% and 36.87% dye are observed with ZnO/Au in presence of UV light. Whereas, for ZnO/Ag catalyst, the amount of dye degradation increased to 68.18% and 42.45%, respectively. This photodegradation result confirms that the photogenerated holes and electrons both can play a major role when catalysis experiments are performed under UV light. The overall scavenging results match well with the earlier literature for both UV and visible light.32,53
Reusability of the catalyst particles
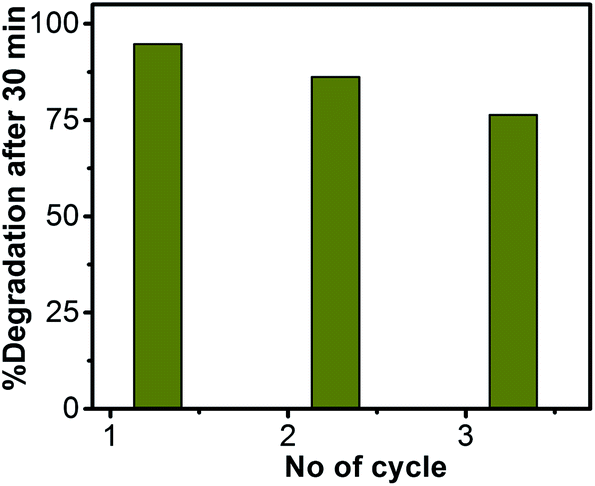
The reusability of ZnO/Au nanoparticles as a photocatalyst in dye decomposition process, have also been tested using a visible light source. The catalyst particles were centrifuged and washed thoroughly before using for each cycle. The photodegradation experiments were repeated for three times with visible light and the corresponding degradation results show 94.7%, 86.2%, and 76.4% of dye degradation for first, second, and third cycle, respectively. Fig. 12 represents the percentage of degradation after 30 min as a function of cycle number, which confirms that the catalysts remain stable even after third cycle. After photocatalysis, the crystal structure of the ZnO, ZnO/Au, and ZnO/Ag catalyst particles was confirmed with the help of PXRD and the result is shown in ESI (Fig. S12†). The reusability of ZnO/Au and ZnO/Ag catalysts using UV light has also been examined and the results confirm its activeness even after third cycle, as can be seen in the ESI, Fig. S13.†
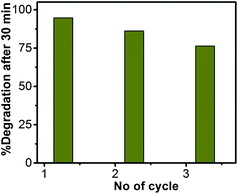 |
| | Fig. 12 Percentage of degradation vs. number of cycle plot for ZnO/Au nanoparticles after 30 min exposure under a visible light source. The catalyst particles were centrifuged and washed well before each cycle of use. | |
Conclusion
In summary, a surfactant assisted simple and convenient method is developed for the preparation of ZnO nanoflower. This ZnO nanoflower surface is further modified with Au and Ag metal nanoparticle which significantly improves the catalytic behaviour of the material. Characterizations accomplish using UV, PL, PXRD, FESEM, TEM, EDS, and XPS techniques very much confirms the formation of nanometer scale Au/Ag dots of homogeneous size and density on top of the flower-like ZnO surfaces. Photodegradation tests using MB dye in presence of UV and visible light ensures a drastic improvement of the catalytic efficiency and photoreactivity of the material after introducing the metal nanoparticles. Scavenging experiments with different scavengers significantly help to understand the underlying mechanism of the reactive species (holes and electrons) involved in photodegradation reaction. Finally, the entire study has a large scale impact and remarkable application on treatment of dye effluents from the textile industries. Therefore, it can be concluded that our investigations can provide a new avenue for the controlled synthesis of size and shape dependent Au and Ag nanoparticles on a semiconductor surface, which can widely be used in electrocatalysis, water splitting etc.
Acknowledgements
SP gratefully acknowledges the financial support from the BITS research initiation grant, University Grant Commission (UGC) start-up-grant (no. F.20-1(21)/2012(BSR)), and Department of Science and Technology (DST) Science and Engineering Research Board (SERB) Fast track (SERB/F/2848/2013-2014) grant. PF gratefully acknowledge to UGC-BSR for financial support. We are also thankful to DST-FIST and UGC-SAP for financial support. The instrumental support for XPS and SEM measurements from Prof. J. Falta of the University of Bremen, Germany is highly acknowledged by SG. We thank to Dr Mrinmoyee Basu (National Taiwan University) for helpful discussions. We also thank to the Department of Physics, BITS Pilani for assistance with powder X-ray diffraction studies (DST-FIST sponsored).
References
-
(a) J. Das and D. Khushalani, J. Phys. Chem. C, 2010, 114, 2544 CrossRef CAS;
(b) A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- J. Becker, K. R. Raghupathi, J. S. Pierre, D. Zhao and R. T. Koodal, J. Phys. Chem. C, 2011, 115, 13844 CAS.
- H. Zeng, W. Cai, P. Liu, X. Xu, H. Zhou, C. Klingshirn and H. Kalt, ACS Nano, 2008, 8, 1661 CrossRef PubMed.
- H. Zeng, P. Liu, W. Cai, S. Yang and X. Xu, J. Phys. Chem. C, 2008, 112, 19620 CAS.
- J. Yuan, E. S. Choo, X. Tang, Y. Sheng, J. Ding and J. Xue, Nanotechnology, 2010, 21, 185606 CrossRef PubMed.
- Y. Zhang, J. Xu, P. Xu, Y. Zhu, X. Chen and W. Yu, Nanotechnology, 2010, 21, 285501 CrossRef PubMed.
- P. Li, Z. Wei, T. Wu, Q. Peng and Y. Li, J. Am. Chem. Soc., 2011, 133, 5660 CrossRef CAS PubMed.
- Q. Wang, B. Geng and S. Wang, Environ. Sci. Technol., 2009, 43, 8968 CrossRef CAS PubMed.
- Y. Zheng, C. Chen, Y. Zhan, X. Lin, Q. Zheng, K. Wei and J. Zhu, J. Phys. Chem. C, 2008, 112, 10773 CAS.
- Q. Deng, X. Duan, D. H. L. Ng, H. Tang, Y. Yang, M. Kong, Z. Wu, W. Cai and G. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 6030 CAS.
- E. Kowalska, H. Remita, C. Colbeau-justin, J. Hupka and J. Belloni, J. Phys. Chem. C, 2008, 112, 1124 CAS.
- P. Wang, B. Huang, Y. Dai and M. H. Whangbo, Phys. Chem. Chem. Phys., 2012, 4, 9813 RSC.
- S. C. Chan and M. A. Barteau, Langmuir, 2005, 21, 5588 CrossRef CAS PubMed.
- H. Zhang, D. Yang, Y. Ji, X. Ma, J. Xu and D. Que, J. Phys. Chem. B, 2004, 108, 3955 CrossRef CAS.
- S. Komarneni, M. Bruno and E. Mariani, Mater. Res. Bull., 2000, 35, 1843 CrossRef CAS.
- T. Trindade, J. D. P. Dejesus and P. O. Bien, J. Mater. Chem., 1994, 4, 1611 RSC.
- X. H. Zhang, S. Y. Xie, Z. Y. Jiang, X. Zhang, Z. Q. Tian, Z. X. Xie, R. B. Huang and L. S. Zheng, J. Phys. Chem. B, 2003, 107, 10114 CrossRef CAS.
- Z. B. Shao, C. Y. Wang, S. D. Geng, X. D. Sun and S. J. Geng, J. Mater. Process. Technol., 2006, 178, 247 CrossRef CAS PubMed.
- O. W. Perez-Lopez, A. C. Farias, N. R. Marcilio and J. M. C. Bueno, Mater. Res. Bull., 2005, 40, 2089 CrossRef CAS PubMed.
- D. Wang and C. Song, J. Phys. Chem. B, 2005, 109, 12697 CrossRef CAS PubMed.
- H. Tang, J. C. Chang, Y. Shan and S. Lee, J. Phys. Chem. B, 2008, 112, 4016 CrossRef CAS PubMed.
- S. Chakraborty, A. K. Kole and P. Kumbhakar, Mater. Lett., 2012, 67, 362 CrossRef CAS PubMed.
- T. T. Miao, D. X. Sun, Y. R. Guo, C. Li, Y. L. Ma, G. Z. Fang and Q. J. Pan, Nanoscale Res. Lett., 2013, 8, 431 CrossRef PubMed.
- K. Kaviyarasu and P. A. Devarajan, Adv. Mater. Lett., 2013, 4, 582 Search PubMed.
- L. Song and S. Zhang, Ind. Eng. Chem. Res., 2012, 51, 4922 CrossRef CAS.
- B. Wen, Y. Huang and J. J. Boland, J. Phys. Chem., 2008, 112, 106 CrossRef CAS PubMed.
- L. Shen, N. Bao, K. Yanagisawa, K. Domen, C. A. Grimes and A. Gupta, J. Phys. Chem. C, 2007, 111, 7280 CAS.
- N. Udawatte, M. Lee, J. Kim and D. Lee, ACS Appl. Mater. Interfaces, 2011, 3, 4531 CAS.
- J. Lee, H. S. Shim, M. Lee, J. K. Song and D. Lee, J. Phys. Chem. Lett., 2011, 2, 2840 CrossRef CAS.
- V. Subramanian, E. E. Wolf and P. V. Kamat, J. Phys. Chem. B, 2003, 107, 7479 CrossRef CAS.
- Y. Zheng, L. Zheng, Y. Zhan, X. Lin, Q. Zheng and K. Wei, Inorg. Chem., 2007, 46, 6980 CrossRef CAS PubMed.
- H. R. Liu, G. X. Shao, J. F. Zhao, Z. X. Zhang, Y. Zhang, J. Liang, X. G. Liu, H. S. B. Jia and S. Xu, J. Phys. Chem. C, 2012, 116, 16182 CAS.
- Z. Zhang, Y. Li, K. Li, K. Chen, Y. Yang, X. Liu, H. Jia and B. Xu, J. Mater. Sci., 2014, 49, 2347 CrossRef CAS.
- A. Mills and J. Wang, J. Photochem. Photobiol., A, 1999, 127, 123 CrossRef CAS.
- S. Pande, S. K. Ghosh, S. Nath, S. Praharaj, S. Jana, S. Panigrahi, S. Basu and T. Pal, J. Colloid Interface Sci., 2006, 299, 421 CrossRef CAS PubMed.
- E. M. Wong and P. C. Searson, Appl. Phys. Lett., 1999, 74, 2939 CrossRef CAS PubMed.
- L. Brus, J. Phys. Chem., 1986, 90, 2555 CrossRef CAS.
- N. Ghobadi, Int. Nano Lett., 2013, 3, 2 CrossRef.
- K. G. Chandrappa and T. V. Venkatesha, Nano-Micro Lett., 2010, 4, 14 Search PubMed.
- S. Sarkar, A. Makhal, T. Bora, S. Baruah, J. Dutta and S. Kumar Pal, Phys. Chem. Chem. Phys., 2011, 13, 12488 RSC.
- A. V. Dijken, E. A. Meulenkamp, D. Vanmaekelbergh and A. Meijerink, J. Phys. Chem. B, 2000, 104, 1715 CrossRef.
- M. Ahmad, S. Yingying, A. Nisar, H. Sun, W. Shen, M. Weie and J. Zhu, J. Mater. Chem., 2011, 21, 7723 RSC.
- L. S. Dake, D. R. Baer and J. M. Zachara, Surf. Interface Anal., 1989, 14, 71 CrossRef CAS.
- S. Pande, S. K. Ghosh, S. Praharaj, S. Panigrahi, S. Basu, S. Jana, A. Pal, T. Tsukuda and T. Pal, J. Phys. Chem. C, 2007, 111, 10806 CAS.
- A. N. Mansour, Surf. Sci. Spectra, 1994, 3, 197 CrossRef CAS.
- M. P. Seah, I. S. Gilmore and G. Beamson, Surf. Interface Anal., 1998, 26, 642 CrossRef CAS.
- S. A. Ansari, M. M. Khan, M. O. Ansari, J. Lee and M. H. Cho, J. Phys. Chem. C, 2013, 117, 27023 CAS.
- R. Saravanan, N. Karthikeyan, V. K. Gupta, E. Thirumal, P. Thangadurai, V. Narayanan and A. Stephen, Mater. Sci. Eng., C, 2013, 33, 2235 CrossRef CAS PubMed.
- P. Sangpour, F. Hashemi and A. Z. Moshfegh, J. Phys. Chem. C, 2010, 114, 13955 CAS.
- J. Liqiang, W. Zhou, G. Tian and H. Fu, Chem. Soc. Rev., 2013, 42, 9509 RSC.
- M. J. Height, S. E. Pratsinis, O. Mekasuwandumrong and P. Praserthdam, Appl. Catal., B, 2006, 63, 305 CrossRef CAS PubMed.
-
(a) H. Li, Z. Bian, J. Zhu, Y. Huo, H. Li and Y. Lu, J. Am. Chem. Soc., 2007, 129, 4538 CrossRef CAS PubMed;
(b) A. Furube, L. Du, K. Hara, R. Katoh and M. Tachiya, J. Am. Chem. Soc., 2007, 129, 14852 CrossRef CAS PubMed.
- N. Zhang, S. Liu, X. Fu and Y. Xu, J. Phys. Chem. C, 2011, 115, 9136 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra03158j |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.