A novel SnS2@graphene nanocable network for high-performance lithium storage†
Debin Kongabc,
Haiyong Heb,
Qi Songb,
Bin Wangb,
Quan-Hong Yang*ac and
Linjie Zhi*abc
aSchool of Chemical Engineering and Technology, Tianjin University, Tianjin, 300072 China. E-mail: zhilj@nanoctr.cn; qhyangcn@tju.edu.cn
bNational Center for Nanoscience and Technology, no. 11 Beiyitiao, Zhongguancun, Beijing, 100190 China
cThe Synergistic Innovation Center of Chemistry and Chemical Engineering of Tianjin, Tianjin, 300072 China
First published on 12th May 2014
Abstract
A unique SnS2@graphene nanocable structure with a novel contact model between SnS2 nanosheets and graphene has been successfully fabricated, in which the graphene layers are rolled up to encapsulate the SnS2 nanosheets, forming a mechanically robust, free-standing SnS2@graphene nanocable network. This distinctive structure provides an effective architecture as an electrode in lithium ion batteries to effectively accommodate the volume change of SnS2 during the charge–discharge cycling, facilitates the easy access of electrolyte to the active electrode materials, and also offers a continuous conductive network for the whole electrode. Interestingly, this binder-free electrode not only shows high specific capacity and excellent cycling performance with a specific capacity of 720 mA h g−1 even after 350 cycles at a current density of 0.2 A g−1 and over 93.5% capacity retention, but exhibits a high-rate capability of 580 mA h g−1 at a current rate of 1 A g−1
Introduction
Rechargeable Li-ion batteries (LIBs) are believed to be one of the most important power sources for portable electronic devices (PED), electric vehicles (EVs) and hybrid electronic vehicles (HEVs).1–3 In order to meet the increasing requirements for PED, EV and HEV applications, intensive research attention has been focused on the development of suitable electrode materials with long cycling life, high rate performance, low cost and enhanced safety for LIBs.4,5 Among these materials, tin disulfides (SnS2) has received particular interest as anode for LIBs due to their high theoretical capacities.6–8 The electrochemical reaction of SnS2 with lithium is proposed to be: (1) SnS2 + 4Li → Sn + 2Li2S, and (2) Sn + xLi ↔ LixSn (x ≤ 4.4).9–11 The first step leads to the first irreversible capacity, while the second step contributes to a reversible theoretical capacity of 645 mA h g−1, significantly higher than that of the commercial graphite anodes (372 mA h g−1).12,13 However, two main drawbacks of this system have stemmed from the low first coulomb efficiency and the large volume change during charge–discharge process, which results in fast capacity decay during electrochemical cycling.14 Although the low first coulombic efficiency can be improved effectively by using Li-rich anodes, for example Li2.6Co0.4N,15 to achieve long cycling life and good rate capability remains a big challenge. Great effort has been made to improve the electrochemical performance of SnS2. For instance, fabricating SnS2 into nanostructured materials, such as fullerene-like nanoparticles,16 nanoflakes,17 nanorods18 and nanobelts19 is beneficial for improving the cycling stability of the electrode. However, to further improve the electrochemical performance with both high-rate capability and high cycling stability, a well-defined conductive network is generally required to accommodate the large volume change of SnS2 and to supply conductive channels.20–23Due to the attractive characteristics such as large specific surface area, superior electronic conductivity and excellent mechanical properties, graphene is considered as an ideal matrix to host various electrochemical active nanoparticles.24–30 Recent research has shown that the combination of graphene with metal oxides,31 Si (ref. 32) and Sn (ref. 33) can significantly enhance the electrochemical properties of these nanostructured anodes. Similar to those of SnO2,34 the electrochemical properties of SnS2 could also be obviously improved when forming SnS2/graphene nanohybrids.12,35,36 Particularly, detailed studies from our group demonstrate the interface effect or the contact model between graphene and SnS2 plays critically important role in improving the rate capability and the cycling stability of the elctrode.13
Herein, we develop a novel nanostructured hybrid composed of SnS2 nanosheets and graphene layers, as schematically shown in Fig. 1a. The graphene layers roll up into a hollow nanotube and SnS2 nanosheets are uniformly distributed in the graphene nanotube, thus forming mechanically robust, free-standing and interwoven SnS2@graphene nanocable (SnS2@GT). The as prepared SnS2@GT can not only effectively alleviate the volume change of SnS2 associated with the alloying/dealloying processes but also facilitate the easy access of electrolyte to the electrodes interior. Interestingly, this nanocable architecture enables a new contact model between graphene and SnS2, providing not only highly efficient interface interaction but a continuous conductive network for the whole electrode. Remarkably, this novel hybrid can be used as binder-free electrode in lithium ion batteries with high specific capacity and excellent cycling performance (720 mA h g−1 even after 350 cycles at a current density of 0.2 A g−1, with over 93.5% capacity retention) as well as high-rate capability of 580 mA h g−1 even at a current rate of 1 A g−1.
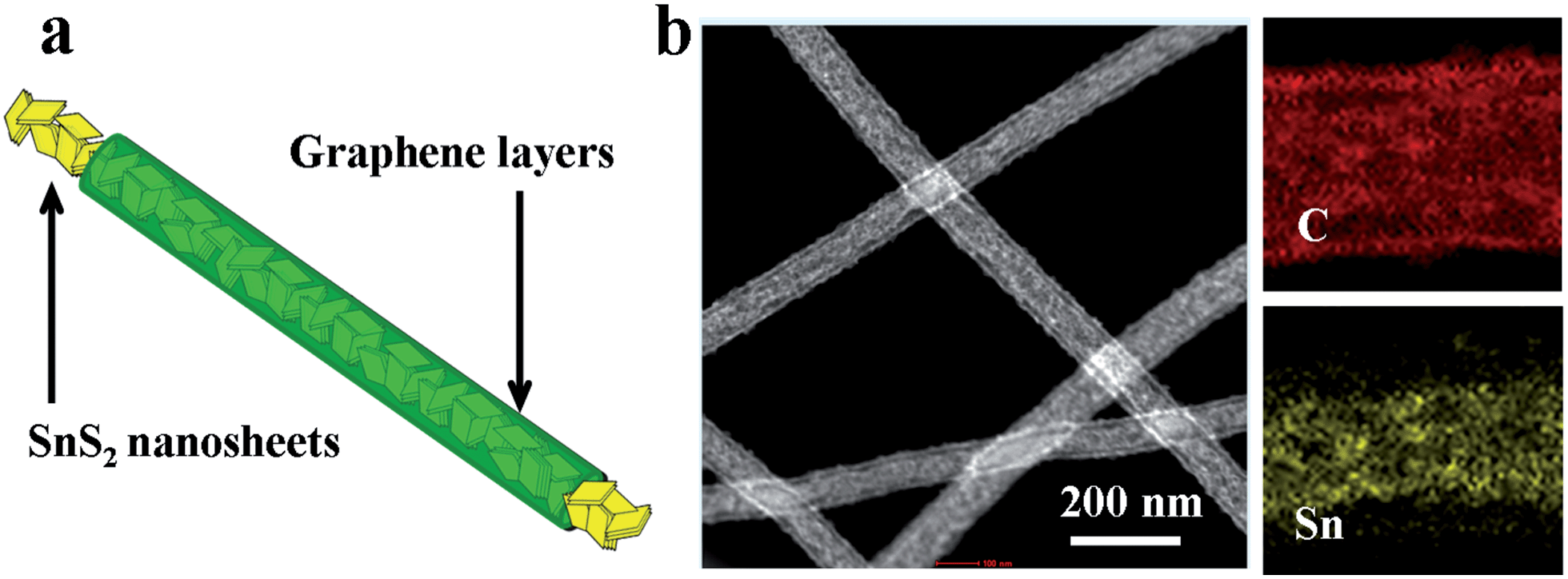 | ||
| Fig. 1 (a) Electrode design and (b) dark field transmission electron microscopy image and mapping of SnS2@GT. | ||
Results and discussion
Field emission scanning electron microscopy (FE-SEM) characterization of a typical SnS2@GT web shows an interconnected nanocable network (Fig. 2a and b). Additionally, each nanocable is around 150 nm in diameter and several micrometers in length. Cross-sectional TEM and HRTEM images (Fig. 3a and b) further demonstrate the nanostructure of a single nanocable, wherein the nanocable wall consists of several graphene layers (average thickness: 10 nm) and the SnS2 nanosheets are evenly encapsulated in a single nanocable. A side view of the SnS2 nanoplates reveals a thickness of about 10 nm (Fig. 3c). The HRTEM image (Fig. 3c and d) exhibits parallel fringes with a spacing of 0.59 nm, which corresponds to the (001) plane of SnS2. Meanwhile, the corrugated parallel fringes with a basal spacing of 0.34 nm corresponds to the (002) plane of graphene layers. Moreover, the size of SnS2 nanosheets is quite small, around tens of nanometers in plane. Elemental mapping results of Sn, S and C elements further proves that ultrathin SnS2 nanosheets distribute evenly along the entire graphene nanocable (Fig. S1 in the ESI†). | ||
| Fig. 2 FE-SEM images of a SnS2@GT web. (a) Scale bar, 5 μm. (b) Scale bar, 2 μm. | ||
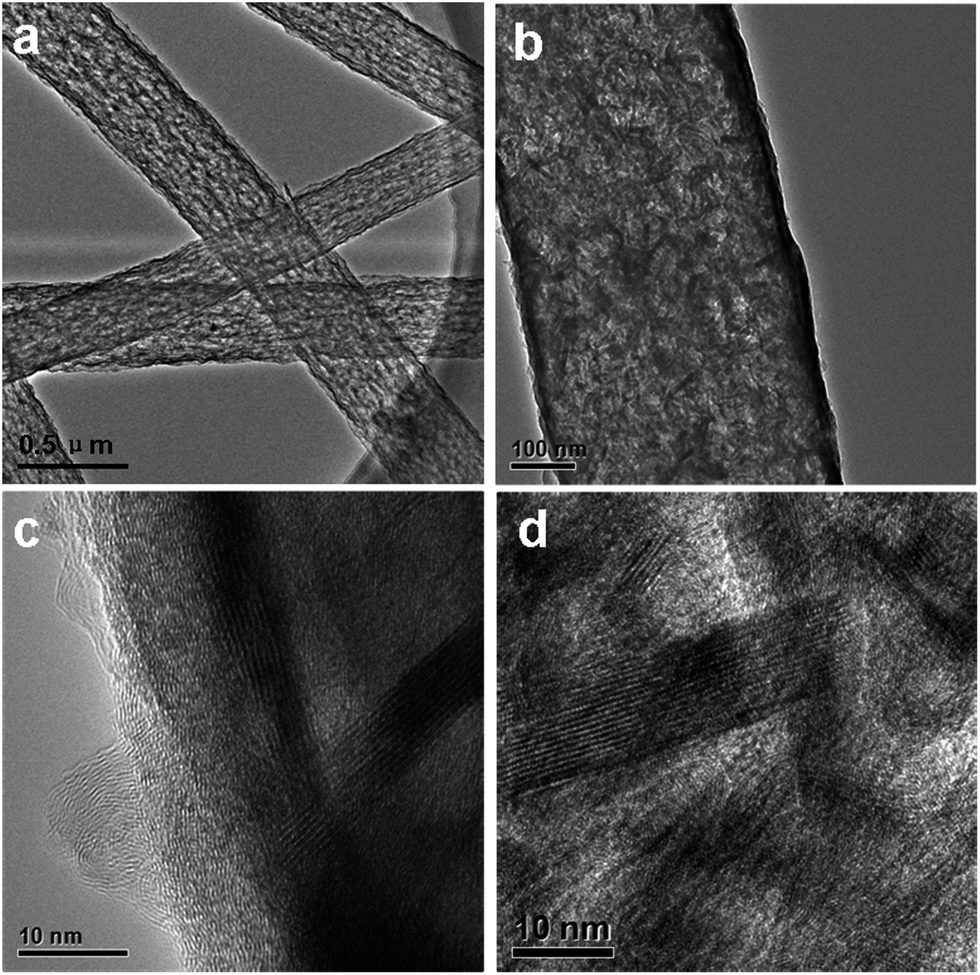 | ||
| Fig. 3 TEM and HRTEM images of SnS2@GT. (a) TEM images of SnS2@GT. Scale bar, 0.5 μm. (b) TEM images of SnS2@GT. Scale bar, 100 nm. (c and d) HRTEM images of SnS2@GT. Scale bar, 10 nm. | ||
The XRD pattern supports the assignment of a layered structure (JCPD card, no. 23-677) to the SnS2 nanoplates (Fig. 4a). No excrescent peak is observed, demonstrating the high purity of as formed SnS2 phase. Besides, the sharp diffraction peak of the (001) plane of SnS2 reveals the good crystallization of the as-synthesized SnS2 nanoparticles in the SnS2@GT. The thickness of the SnS2 nanoplates is estimated to be around 10 nm, using the Scherrer equation37 according to the (001) peak, which is complying with the estimated thickness from TEM. The presence of both SnS2 and graphene is further confirmed by Raman spectroscopy (Fig. 4b). The Raman spectrum of SnS2@GT exhibits an intense peak at about 311 cm−1, which is attributed to the A1g mode of SnS2 according to the group theory analysis conducted by previous studies.38 The peak at about 1585 cm−1 (G band), corresponding to an E2g mode of graphite, is related to the vibration of the sp2-bonded carbon atoms in a two dimensional hexagonal lattice, while the peak at about 1325 cm−1 (D band) is related to the defects and disorder in the hexagonal graphitic layers.39
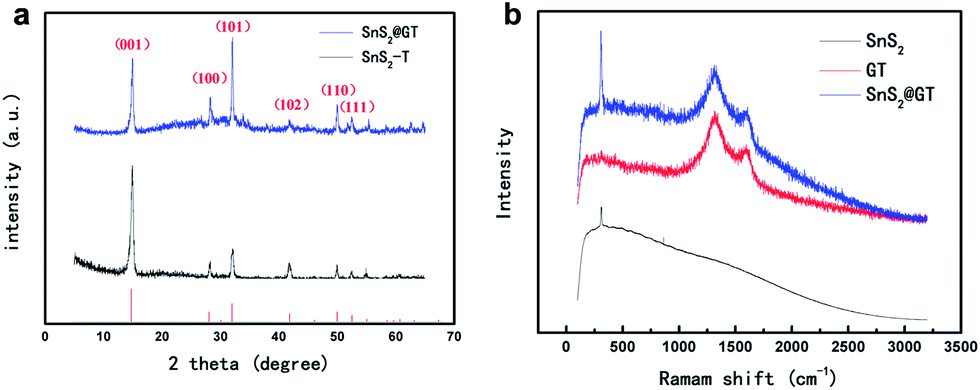 | ||
| Fig. 4 (a) XRD patterns of SnS2@GT. (b) Raman patterns of SnS2@GT. | ||
Based on the unique nanocable architecture, the SnS2@GT hybrid has potential applications in the context of lithium ion storage. For comparison, bare SnS2 (SnS2-T) were prepared via a similar route as that of nanocable formation (see the Experimental section) and investigated under the same electrochemical conditions. The cyclic voltammetry (CV) behavior (Fig. S2†) of SnS2@GT is generally consistent with that of the SnS2 reported previously,40,41 indicating similar electrochemical reaction pathway of the two electrode materials. Fig. 5a and b compare the voltage profiles between SnS2@GT and SnS2-T charged–discharged at 0.2 A g−1 in the cutoff voltage of 0.01–3.0 V vs. Li/Li+. As shown in the Fig. 5a, SnS2@GT yields the first discharge and charge capacities of 1334 and 764 mA h g−1, respectively, with a coulomb efficiency of 58.3%, while the first charge and discharge capacities of bare SnS2-T are 1075 and 335 mA h g−1, respectively, with the Coulomb efficiency of only 31.2%.
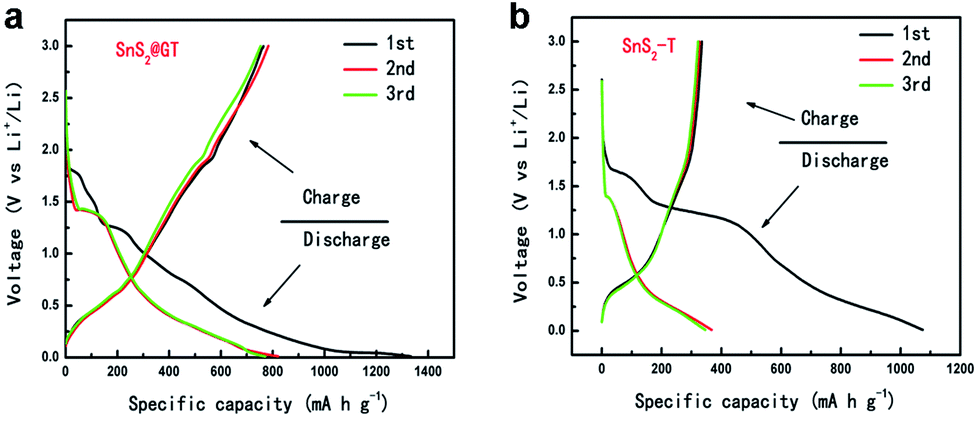 | ||
| Fig. 5 The charge–discharge curves of (a) SnS2@GT and (b) SnS2-T electrode at a current rate of 0.2 A g−1. | ||
As seen in Fig. 5b, most of the charge capacity of bare SnS2-T is obtained in the voltage range of 0.01–1.5 V, where lithiation and delithiation of Sn occur. The voltage increases rapidly to above 1.5 V, which indicates that voltage beyond 1.5 V only contributes a small proportion of the total capacity and that the lithium ions in Li2S is difficult to be extracted upon charging, similar to the case of SnO2, where the formation of Li2O during first discharge is also irreversible.42
For the SnS2@GT, the initial discharge capacity is slightly higher than the theoretical values reported previously. It is possible that the high surface area of the SnS2 nanoparticles, which may have promoted side-reactions with electrolyte and in the meantime intercalate the lithium ions into the SnS2 layers without phase decomposition at 1.8 V, is responsible for the high initial discharge capacity. Fig. 6a shows typical charge–discharge curves of the SnS2@GT and SnS2-T electrodes cycled between 3 V and 0.01 V. The SnS2@GT electrode clearly demonstrates its superior cycling performance and much better long-term stability than that of the SnS2-T electrode. The coulombic efficiency for SnS2@GT (the content of SnS2 is 71.6%, as confirmed by TGA in Fig. S3†) is 58% in the initial cycle, which rapidly reaches over 95% after the second cycle. Even after 350 cycles, both the discharge and charge capacities of this material are stable at about 720 mA h g−1, delivering nearly 92% capacity retention, whereas the SnS2-T electrode shows poor cycling performance with an initial reversible capacity of merely 335 mA h g−1, followed by a gradual reduction to 24 mA h g−1 after 170 cycles, which corresponds to merely 7.2% of the reversible capacity obtained in the first cycle. The results clearly demonstrate that the induced graphene nanotubes can effectively accommodate the volume change of SnS2, resulting in greatly enhanced cycling stability.
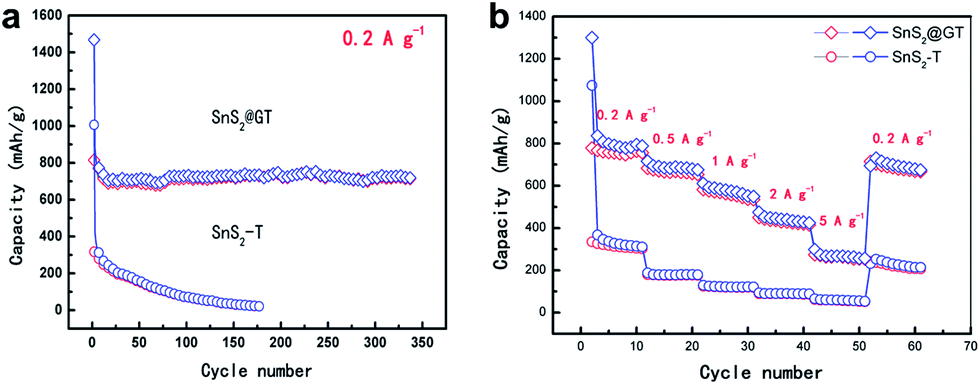 | ||
| Fig. 6 Electrochemical performance of SnS2@GT and SnS2-T electrodes. (a) Electrochemical cycling performance of SnS2@GT and SnS2-T electrodes under charge–discharge cycles from 0.01 to 3.0 V. (b) Reversible capacity of SnS2@GT and SnS2-T at various current rates from 0.2 to 5 A g−1. | ||
The rate capability of SnS2@GT and SnS2-T is evaluated to further investigate the effect of graphene incorporation on the electrochemical performance of SnS2 (Fig. 6b). The samples were charged at various current densities and discharged at 0.2 A g−1. It is obvious that the SnS2@GT hybrid displays a much better rate capability than SnS2-T. When the current density reaches 1 A g−1, the SnS2@GT electrode can still retain a charge capacity of 580 mA h g−1. Even at a current density as high as 5 A g−1, SnS2@GT still yields an attractive charge capacity of 247 mA h g−1. By contrast, the charge capacities of bare SnS2-T drop dramatically with increasing the current density.
To gain an insight view for the reason of such excellent performance of SnS2@GT architecture, we conducted electrochemical impedance measurements on SnS2@GT and SnS2-T to reveal their electrochemical reaction kinetics (Fig. S4†). Both plots are characteristic of one semicircle in the high frequency region and a straight sloping line in the low frequency region. In general, the semicircle is attributed to the summation of the contact resistance, the solid/electrolyte interphase resistance, and the charge-transfer resistance, while the slope of the line is closely related to the lithium-diffusion process within the electrodes. Apparently, compared to SnS2-T electrode, SnS2@GT electrodes hold much smaller semicircle diameter and almost a vertical line parallel to the Zireal as well. This is clear evidence that the SnS2@GT possesses a higher electrical conductivity and a faster charge-transfer reaction for lithium ion insertion and extraction.
Compared with other previously reported SnS2/graphene hybrids, the as prepared SnS2@GT sample exhibits an improved rate capability and good capacity retention under similar measurement conditions,10,13,22,41 which possibly could be attributed to its multi-scale structure and hierarchical features. Firstly, in such structure, the flexible film constructed by the interconnection of nanocables provides the most efficient electron transport pathways and forms a strong skeleton framework. Secondly, the induced graphene nanotubes with good mechanical properties can facile strain relaxation and accommodate the volume change of SnS2, which effectively mitigates the stress and protects active materials from pulverization during the charge–discharge process. Thirdly, the small size of SnS2 nanosheets decreases the over voltage of the Sn–Li alloying reaction, leading to a higher discharge capacity. At the beginning of the discharging process, it occurs mainly at the external surface of the electrode, which is favorable for the fast transport of lithium ions and electrons. However, during the subsequent cycles, the electrode reaction will move gradually into the interior of the electrode, leading to the increase of diffusion over potential. Obviously, the small size of SnS2 nanosheets is favorable for decreasing the over voltage of the Sn–Li alloying reaction, leading to a higher discharge capacity.
Experimental
General procedure for the synthesis of SnS2@GT
First, 0.32 g tin(II) chloride dihydrate (SnCl2) is dissolved in 2 mL DMF. A flexible film composed by SnCl2–SiO2–PVP is produced through electrospinning of 2 mL DMF solution of 0.32 g PVP with the addition of 2 mL ethanol solution and 640 μL TEOS. Afterward, the flexible film obtained by electrospinning was heated to 1070 °C at a rate of 10 °C min−1 in a tube furnace under argon (Ar) atmosphere. Then, 100 sccm methane (CH4) was introduced into the reaction tube and kept for 5 min. After that, the sample was rapidly cooled done to room temperature under the protection of Ar, thus obtaining a textured SnS2@GT film followed by a mild oxidation at 350 °C. Subsequently, SnS2@GT is finally obtained by removing SiO2 with HF and then annealing at 400 °C for 4 h in H2S.General procedure for the synthesis of SnS2-T
SnS2-T is prepared with a method similar to that of SnS2@GT, excepting that no CH4 is available during the CVD process and without the removal of SiO2.Characterization
The morphologies of the samples were examined by the scanning electron microscope (Hitachi S4800) and field emission transmission electron microscopy (FEI Tecnai G2 20 ST). Nitrogen adsorption isotherms of the samples were measured with a Micromeritics ASAP2020 instrument, and the surface area was obtained by Brunauer–Emmett–Teller (BET) analyses. Pore size distributions were calculated by using the Barret–Joyner–Halenda (BJH) model based on nitrogen desorption isotherm. X-ray diffraction (XRD) with Cu Kα radiation (Rigaku D/max-2500B2+/PCX system) was used to determine the phase composition and the crystallinity.Electrochemical characterizations
Both the free-standing SnS2@GT and SnS2-T films were directly used as the working electrode. The as-made working electrodes were assembled into coin-type half cells (CR2032) in an argon-filled glovebox (<1 ppm of oxygen and water) with lithium foil as the counter electrode, porous polypropylene film as the separator, and 1 M LiPF6 in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) ethylene carbonate/diethyl carbonate (EC/DEC) as the electrolyte. The cycle-life tests were performed using a CT2001A battery program controlling test system at different current rates within the 3–0.01 V voltage range. For achieving the capacity values of each electrode material, at least three cells were assembled and characterized under the same conditions. For each investigated electrode, the total electrode weight was used for calculating the specific capacities.
:1 (v/v) ethylene carbonate/diethyl carbonate (EC/DEC) as the electrolyte. The cycle-life tests were performed using a CT2001A battery program controlling test system at different current rates within the 3–0.01 V voltage range. For achieving the capacity values of each electrode material, at least three cells were assembled and characterized under the same conditions. For each investigated electrode, the total electrode weight was used for calculating the specific capacities.
Conclusions
In summary, we have successfully designed and prepared a novel SnS2@graphene nanocable network, in which the graphene are rolled up to encapsulate SnS2 nanosheets into a cable structure, thus forming a free-standing, mechanically robust, interwoven SnS2@GT nanocable web. Such a unique architecture can accommodate effectively the volume change of SnS2 during charge–discharge cycling, facilitate the easy access of electrolyte to the active electrode materials, and provide as well a continuous conductive network for the whole electrode. When being used as a binder-free anode on the basis of the whole electrode, this hybrid material not only shows high specific capacity and excellent cycling performance (720 mA h g−1 even after 350 cycles at a current density of 0.2 A g−1, with over 93.5% capacity retention) but exhibits a high-rate capability of 580 mA h g−1 even at the current rate of 1 A g−1. In addition, the strategy developed here can also be used to fabricate various 2D/2D hybrids materials for potential applications in LIBs, supercapacitors, and catalysis.Acknowledgements
The authors would like to thank Bin Luo and Lin Shi for their help and discussions and the finance supports from the Ministry of Science and Technology of China (nos 2012CB933403 and 2014CB932403), the National Natural Science Foundation of China (Grant nos 20973044, 21173057 and 51372167), and the Chinese Academy of Sciences.Notes and references
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS PubMed.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- A. S. Aricò, P. Bruce, B. Scrosati, J.-M. Tarascon and W. Van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef PubMed.
- M. Wachtler, M. Winter and J. O. Besenhard, J. Power Sources, 2002, 105, 151–160 CrossRef CAS.
- I. A. Courtney and J. Dahn, J. Electrochem. Soc., 1997, 144, 2943–2948 CrossRef CAS PubMed.
- J. Zai, K. Wang, Y. Su, X. Qian and J. Chen, J. Power Sources, 2011, 196, 3650–3654 CrossRef CAS PubMed.
- C. Zhai, N. Du and H. Z. D. Yang, Chem. Commun., 2011, 47, 1270–1272 RSC.
- T.-J. Kim, C. Kim, D. Son, M. Choi and B. Park, J. Power Sources, 2007, 167, 529–535 CrossRef CAS PubMed.
- H. S. Kim, Y. H. Chung, S. H. Kang and Y.-E. Sung, Electrochim. Acta, 2009, 54, 3606–3610 CrossRef CAS PubMed.
- H. Zhong, G. Yang, H. Song, Q. Liao, H. Cui, P. Shen and C.-X. Wang, J. Phys. Chem. C, 2012, 116, 9319–9326 CAS.
- S. Liu, X. Lu, J. Xie, G. Cao, T. Zhu and X. Zhao, ACS Appl. Mater. Interfaces, 2013, 5, 1588–1595 CAS.
- B. Luo, Y. Fang, B. Wang, J. Zhou, H. Song and L. Zhi, Energy Environ. Sci., 2012, 5, 5226–5230 CAS.
- Y. Idota, T. Kubota, A. Matsufuji, Y. Maekawa and T. Miyasaka, Science, 1997, 276, 1395–1397 CrossRef CAS.
- J. Yang, Y. Takeda, N. Imanishi and O. Yamamoto, J. Electrochem. Soc., 2000, 147, 1671–1676 CrossRef CAS PubMed.
- S. Y. Hong, R. Popovitz-Biro, Y. Prior and R. Tenne, J. Am. Chem. Soc., 2003, 125, 10470–10474 CrossRef CAS PubMed.
- D. Chen, G. Shen, K. Tang, S. Lei, H. Zheng and Y. Qian, J. Cryst. Growth, 2004, 260, 469–474 CrossRef CAS PubMed.
- X.-L. Gou, J. Chen and P.-W. Shen, Mater. Chem. Phys., 2005, 93, 557–566 CrossRef CAS PubMed.
- Y. Ji, H. Zhang, X. Ma, J. Xu and D. Yang, J. Phys.: Condens. Matter, 2003, 15, L661 CrossRef CAS.
- C. Liu, F. Li, L. P. Ma and H. M. Cheng, Adv. Mater., 2010, 22, E28–E62 CrossRef CAS PubMed.
- G. Derrien, J. Hassoun, S. Panero and B. Scrosati, Adv. Mater., 2007, 19, 2336–2340 CrossRef CAS.
- P. Chen, Y. Su, H. Liu and Y. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 12073–12082 CAS.
- M. Sathish, S. Mitani, T. Tomai and I. Honma, J. Phys. Chem. C, 2012, 116, 12475–12481 CAS.
- Z. Fan, J. Yan, L. Zhi, Q. Zhang, T. Wei, J. Feng, M. Zhang, W. Qian and F. Wei, Adv. Mater., 2010, 22, 3723–3728 CrossRef CAS PubMed.
- D. Wang, R. Kou, D. Choi, Z. Yang, Z. Nie, J. Li, L. V. Saraf, D. Hu, J. Zhang and G. L. Graff, ACS Nano, 2010, 4, 1587–1595 CrossRef CAS PubMed.
- S. Yang, X. Feng, L. Wang, K. Tang, J. Maier and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 4795–4799 CrossRef CAS PubMed.
- Y. Gong, S. Yang, Z. Liu, L. Ma, R. Vajtai and P. M. Ajayan, Adv. Mater., 2013, 25, 3979–3984 CrossRef CAS PubMed.
- K. Chang and W. Chen, Chem. Commun., 2011, 47, 4252–4254 RSC.
- K. Chang and W. Chen, ACS Nano, 2011, 5, 4720–4728 CrossRef CAS PubMed.
- S. Yang, G. Cui, S. Pang, Q. Cao, U. Kolb, X. Feng, J. Maier and K. Müllen, ChemSusChem, 2010, 3, 236–239 CrossRef CAS PubMed.
- Z.-S. Wu, G. Zhou, L.-C. Yin, W. Ren, F. Li and H.-M. Cheng, Nano Energy, 2012, 1, 107–131 CrossRef CAS PubMed.
- B. Wang, X. Li, T. Qiu, B. Luo, J. Ning, J. Li, X. Zhang, M. Liang and L. Zhi, Nano Lett., 2013, 13, 5578–5584 CrossRef CAS PubMed.
- B. Luo, B. Wang, X. Li, Y. Jia, M. Liang and L. Zhi, Adv. Mater., 2012, 24, 3538–3543 CrossRef CAS PubMed.
- S.-M. Paek, E. Yoo and I. Honma, Nano Lett., 2008, 9, 72–75 CrossRef PubMed.
- J. Yin, H. Cao, Z. Zhou, J. Zhang and M. Qu, J. Mater. Chem., 2012, 22, 23963–23970 RSC.
- L. Zhuo, Y. Wu, L. Wang, Y. Yu, X. Zhang and F. Zhao, RSC Adv., 2012, 2, 5084–5087 RSC.
- A. Patterson, Phys. Rev., 1939, 56, 978 CrossRef CAS.
- C. Wang, K. Tang, Q. Yang and Y. Qian, Chem. Phys. Lett., 2002, 357, 371–375 CrossRef CAS.
- A. Ferrari, J. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. Novoselov and S. Roth, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS.
- Q. Wu, L. Jiao, J. Du, J. Yang, L. Guo, Y. Liu, Y. Wang and H. Yuan, J. Power Sources, 2013, 239, 89–93 CrossRef CAS PubMed.
- J. W. Seo, J. T. Jang, S. W. Park, C. Kim, B. Park and J. Cheon, Adv. Mater., 2008, 20, 4269–4273 CrossRef CAS.
- I. A. Courtney and J. Dahn, J. Electrochem. Soc., 1997, 144, 2045–2052 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra03052d |
| This journal is © The Royal Society of Chemistry 2014 |