
Hydroxyl and ester functionalized N-heterocyclic carbene complexes of iridium(i): efficient catalysts for transfer hydrogenation reactions†
Abstract
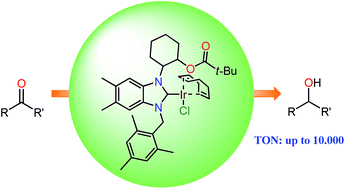
Hydroxyl and ester-functionalized iridium(I) complexes of N-heterocyclic carbenes (3a–e) were obtained by transmetalation reactions from the in situ prepared silver(I)–NHC complexes and characterized by IR, NMR, mass spectroscopies, and elemental analysis. X-ray diffraction studies on single crystals of 3a, 3c and 3d verify the square planar geometry at the iridium center. Ester functionalized iridium(I)–NHC complexes were found to be highly active and selective catalysts for the transfer hydrogenation reactions of various aldehydes and ketones. The influence of different ester substituents on the reactivity of the complexes was studied and the complex with a pivaloyl substituent (3d) showed the best activity (TON up to 10 000).
 Please wait while we load your content...
Please wait while we load your content...

