
A fluorescence assay for the trace detection of protamine and heparin†
Abstract
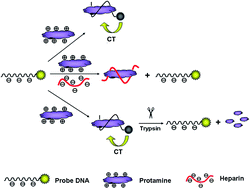
Here we designed a fluorescence sensing strategy for the determination of protamine, heparin and trypsin. A fluorescein isothiocyanate (FITC)-labeled DNA probe could wrap around protamine molecules tightly via electrostatic interactions accompanied by the fluorescence quenching of the DNA probe. In the presence of heparin, protamine preferred to bind to heparin instead of DNA due to the stronger affinity of heparin to protamine, and the fluorescence could be restored. Based on the fact that the protamine which is rich in arginine could be hydrolyzed by the addition of trypsin, the fluorescence intensity of the DNA/protamine complex could also be linked to the trypsin concentration. By measuring the fluorescence intensity changes, the concentrations of protamine, heparin and trypsin were determined. Under the optimized conditions, the linear response range was obtained from 2.5 to 17.5 ng mL−1, 0.156–1.875 ng mL−1 and 62.5 to 10 000 ng mL−1 with the low detection limit of 2.2 ng mL−1, 0.078 ng mL−1 and 30.2 ng mL−1 for protamine, heparin and trypsin, respectively.
 Please wait while we load your content...
Please wait while we load your content...

