
Effect of CuMn2O4 spinel in Cu–Mn oxide catalysts on selective catalytic reduction of NOx with NH3 at low temperature
Abstract
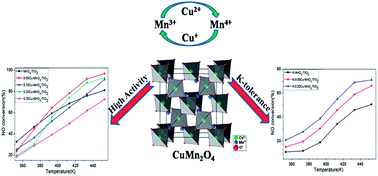
Using the impregnation method, a series of Cu–Mn oxide catalysts were prepared and investigated for the selective catalytic reduction (SCR) of NOx with NH3 at temperatures ranging from 353 K to 453 K. The 0.05Cu–MnOx/TiO2 catalyst shows the highest activity and yields nearly 100% NOx conversion at 453 K using GHSV = 40 000 h−1, while the 0.20Cu–MnOx/TiO2 catalyst exhibits a certain level of potassium tolerance. In addition, the catalysts show favorable stability and water resistance. According to the XRD, EDS and SCR performance results, the existence of a new crystallized CuMn2O4 spinel phase is the dominant parameter for outstanding SCR activity between 413 K and 453 K. TPR, XPS and in situ DRIFT experiments indicate that CuMn2O4 is responsible for low reduction temperature, strong interaction between manganese oxides and copper oxides, high Mn3+ content and numerous acid sites on the surface. Compared with MnOx/TiO2 catalysts, Cu–Mn oxide catalysts could reduce the poisoning effect of potassium, illustrating that the CuMn2O4 phase may play a significant role in K-tolerance. Meanwhile, based on a certain level of potassium tolerance in CuMn2O4, an oxidation mechanism for NO is proposed due to the increase in Mn3+ and the special structure of a spinel oxide.
 Please wait while we load your content...
Please wait while we load your content...

