
Dimerization of heteroaromatic N-oxides under metal-free conditions†
Abstract
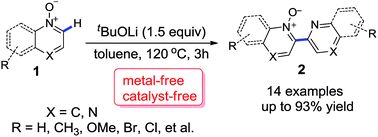
A novel procedure for the dimerization of heteroaromatic N-oxides under transition-metal-free conditions has been developed. The protocol is effective and convenient. The biheteroaromatic mono N-oxides products were obtained in up to 93% yield.
 Please wait while we load your content...
Please wait while we load your content...

