Crystalline Li3V6O16 rods as high-capacity anode materials for aqueous rechargeable lithium batteries (ARLB)†
Vivek Sahadevan Nair‡ab,
Sivaramapanicker Sreejith‡c,
Parijat Borahc,
Steffen Hartungde,
Nicolas Bucherde,
Yanli Zhaoac and
Srinivasan Madhavi*abd
aSchool of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, 639798, Singapore. E-mail: madhavi@ntu.edu.sg
bInstitute for Sports Research, Nanyang Technological University, 50 Nanyang Avenue, 639798, Singapore
cDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, 637371, Singapore
dTUM-CREATE, 1-Create Way, 138602, Singapore
eTechnische Universität München, 85748, Garching, Germany
First published on 30th April 2014
Abstract
We report the preparation of highly crystalline Li3V6O16 rods (LVO-1 rods) and their use as anode materials in aqueous rechargeable lithium ion batteries (ARLBs) for the first time. Half-cell ARLBs with LVO-1 as an anode material deliver a higher initial capacity of >120 mA h g−1 at high current rates and a higher round trip efficiency of 52% at the end of 100 cycles at a current density of 500 mA g−1. ARLBs with LVO-1 rods as anodes exhibited an excellent rate and cycling performance.
Nowadays, most portable electronic devices use lithium ion batteries as their major source of energy storage.1–6 Major problems related to lithium ion batteries include the use of flammable organic electrolytes, the presence of highly reactive and inflammable electrode components and difficulty in their usage.7–9 Also, leaks in the packaging of a lithium ion battery can cause the battery to combust and prevent it from performing. Most of the major fires and accidents in automobile and aircraft engines are due to lithium ion batteries.10 Recently, three Tesla Motors Model S electric cars caught fire after their lithium ion battery packs were damaged, raising doubts in the minds of the public regarding safety and classing electric vehicles as fire hazards.11 Hence, it would be even more difficult to imagine using lithium ion batteries in wearable sports applications, where the batteries come in direct contact with the human body.
Li et al.12 reported a new possibility for energy storage i.e., aqueous rechargeable lithium ion batteries (ARLBs), which are a safer alternative and could prove to be competent enough to match the performance of current lithium ion batteries. As ARLB systems use water as the electrolyte medium, leakages in an ARLB package would most probably not result in a fire or reduce the performance of the battery, thus rendering it useful for many applications like electric vehicles, large scale grid based energy storage and more importantly, in portable and wearable electronics.13,14 However, owing to the complicated reaction possibilities in a typical ARLB, a rational selection of the electrode materials used in ARLBs remains challenging.13–16 Only electrodes which fall within the oxidation and reduction potential of water, avoiding the release of oxygen or hydrogen during the battery charge–discharge process, can be used for any practical application. Hence, the overall voltage, and therefore energy density, of the battery is limited due to the nature of an electrode that operates in an aqueous system.17 An increase in the overall energy density is detrimental when attempting to make ARLBs equally competent to lithium ion batteries. In this context, the design of new electrode materials for ARLBs with higher rates of performance is of great importance.
Generally in ARLB systems, LiMn2O4 has been studied extensively with an operating voltage of 0–1.0 V vs. SCE (standard calomel electrode).18 Commonly used anode materials in ARLBs, like TiP2O7, LiTi2(PO4)3 and LiV3O8, give discharge capacities of 100 mA h g−1, 80 mA h g−1 and 40 mA h g−1 respectively, with cycle life efficiencies of only ∼35% (25 cycles), ∼38% (25 cycles) and 53.5% (100 cycles) respectively.15,16,19 Vanadium oxides and bronzes are sought out to be the best materials for large scale energy storage due to their tunable structural flexibility, high discharge capacity, abundance and low cost amongst the numerous other transition metal oxides.20–24 Various vanadium oxides like LiV2O5, LiV3O8, and VO2 (B) have been studied for ARLB applications but their practical capacities are 47 mA h g−1, 45 mA h g−1 and 100 mA h g−1 respectively, and their capacities fall down to approximately <50% at the end of 100 cycles.25 In the case of LiV3O8, its layers are made up of VO6 octahedra and VO5 tetrahedra which are held together by immobile lithium ions situated at the interlayer octahedral sites. In aqueous electrolytes, LiV3O8 shows a capacity of <45 mA h g−1 and a poor cycling performance due to vanadium dissolution followed by the collapse of its crystal structure. To improve the overall efficiency and usability of ARLBs, the development of a potential anode material with a sufficiently high capacity, higher voltage window and increased cycle life stability is essential.
Here, we report a facile hydrothermal preparation of Li3V6O16 rods (hereafter referred to as LVO-1 rods) with high crystallinity and demonstrated their application as anode materials in ARLBs. In this work, we report a novel Li3V6O16 anode material with a wide operating potential, −1.1–0 V vs. SCE, that can be used as an anode to fabricate a 2 V ARLB system with cathode materials like LiMn2O4, LiNiO2, LiCoO2, LiFePO4, thereby improving the overall energy density of the ARLB system significantly. In a typical experiment, a mixture of lithium acetate (CH3COOLi·2H2O) and vanadium pentoxide (V2O5) was prepared in 25 mL distilled deionized water in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2 molar ratio. The resulting solution was mixed thoroughly by stirring until it turned red with a pH of 9.0. The solution was neutralized and allowed to undergo a hydrothermal process in a Teflon-lined stainless steel autoclave for 48 h at 200 °C. The product was collected by centrifugation, followed by repeated washing in deionized water and ethanol in order to remove any possible ionic remnants and dried in a vacuum overnight at 105 °C. The sample was then calcinated at 400 °C and ground in order to obtain the highly crystalline LVO-1.
:1.2 molar ratio. The resulting solution was mixed thoroughly by stirring until it turned red with a pH of 9.0. The solution was neutralized and allowed to undergo a hydrothermal process in a Teflon-lined stainless steel autoclave for 48 h at 200 °C. The product was collected by centrifugation, followed by repeated washing in deionized water and ethanol in order to remove any possible ionic remnants and dried in a vacuum overnight at 105 °C. The sample was then calcinated at 400 °C and ground in order to obtain the highly crystalline LVO-1.
The readily prepared LVO-1 was then characterized using field emission scanning electron microscopy (FE-SEM), high resolution transmission electron microscopy (HR-TEM) and atomic force microscopy (AFM). An FE-SEM image (Fig. 1a) of LVO-1 shows extended rod-like morphology with an aspect ratio of ∼20 (average diameter of ∼0.5 μm and length ≤5 μm). The TEM image shown in Fig. 1b shows clear formation of extended rods. From the HR-TEM image (Fig. 1c) the obtained lattice fringe pattern (Fig. 1d) and selected area electron diffraction (Fig. 1e) of the LVO-1 rods reveal the presence of several layers of lamellar crystals, which lead to the formation of an extended rod morphology. LVO-1 has a higher surface area thereby rendering it useful for faster charge–discharge in batteries. Similarly, the AFM image (Fig. 1f) also shows the rod-like morphology of LVO-1 and is consistent with the images observed by FE-SEM and TEM. Powder XRD patterns (Fig. S1†) were collected to elucidate the crystal structure of LVO-1 and evaluated using the Rietveld refinement method. The XRD patterns (Fig. S1†) can be fully indexed to the crystal structure of Li3V6O16 (P121/m1, PDF 4-0417) via Rietveld refinement (R factor <0.06), without the presence of any impurity peaks. The XRD data shown in Fig. S1† for LVO-1 can be reasonably indexed to the observed selected area diffraction pattern in Fig. 1e.
 | ||
| Fig. 1 LVO-1: (a) FE-SEM image, (b) TEM image (low resolution), (c) HR-TEM image, and (d) lattice fringe obtained from the selected area in (c). (e) SAED pattern and (f) AFM image. | ||
X-ray Photoelectron Spectroscopy (XPS) survey spectra (ESI, Fig. S2†) reveal the presence of lithium, vanadium and oxygen without any impurities. The high resolution spectra of V2p3/2 and V2p1/2 centered at 516.1 and 523.6 eV respectively, indicate the presence of V5+ species (Fig. S3a†). Similarly for the Li 1s region, the core level is at 54.85 eV, with the dominant Li signal assigned to Li+ (Fig. S3b†). Hence we proved the monoclinic nature of Li3V6O16 with no evident impurities detected.
To elucidate the electrochemical processes of the ARLBs, cyclic voltammetry (CV) and galvanostatic charge–discharge cycling of the half-cells were carried out in a three-electrode configuration. A 3 M lithium nitrate (LiNO3) aqueous solution was used as the electrolyte, with platinum foil and a SCE as the counter and reference electrodes respectively. The voltage range was computer-controlled using a potentiostat (Solartron, 1470E) at room temperature, in the voltage range of 0 to 1.9 V vs. SCE. In order to investigate the phase transformations occurring during the electrochemical processes, cyclic voltammetric tests (Fig. 2) of the LVO-1 rods as anodes were carried out in a three-electrode configuration, within a potential range of −1.1 to 0.0 V at scan rates of 0.5 mV s−1 and 2 mV s−1. In the anodic cyclic voltammogram of LVO-1, two main reduction peaks can be observed at −0.4 V and −0.6 V, while a faint reduction peak can be observed at −0.3 V vs. SCE and two main oxidation peaks at −0.425 V and −0.275 V vs. SCE, respectively. These peaks indicate lithium insertion (reduction)–de-insertion (oxidation) processes occurring during electrochemical charging–discharging.
 | ||
| Fig. 2 Cyclic voltammograms of LVO-1 as an anode in a three electron configuration (vs. SCE) from −1.2 to 0 V at scan rates of 0.5 mV s−1 and 2 mV s−1. | ||
We then evaluated the electrochemical charge–discharge performance of LVO-1 as an anode, as shown in Fig. 3a, using a three-electrode configuration at a current density of 0.5 mA h g−1. The LVO-1 anode gave an initial charge capacity of 110 mA h g−1 and a discharge capacity of 120 mA h g−1. All the specific capacity values were calculated with respect to the mass of the active material. LVO-1 displays a slightly higher anode capacity at a higher negative voltage window compared to other commonly used anode materials in ARLBs, like TiP2O7, LiT2(PO4)3 and LiV3O8. The absence of a flat discharge plateau can be attributed to the multi-lithium ion intercalation–de-intercalation process.25 The cyclic performance of LVO-1 active materials was further evaluated by prolonged cycling, up to 100 cycles at 0.5 Ag−1, as shown in Fig. 3b. Fig. 3c shows the cyclic stability of LVO-1 for 10 cycles, each at varying current densities. The discharge capacity at the end of 100 cycles for LVO-1 was 62.5 mA h g−1 with ∼52% equivalent to retention of the initial capacity. Hence, the cyclic stability of LVO-1 can be attributed to the higher surface area and higher crystallinity (larger crystal size) as evident from the TEM, AFM (Fig. 1) and XRD results (Fig. S1†).
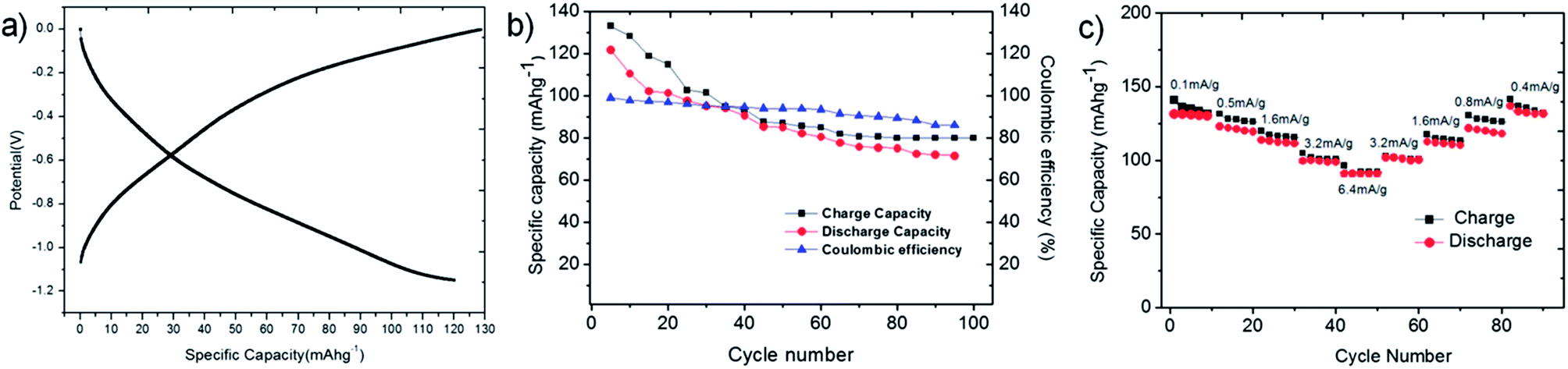 | ||
| Fig. 3 (a) Galvanostatic discharge–charge profiles of LVO-1 in 3 M LiNO3 aqueous solution, in a three-electrode configuration (vs. SCE) from −1.2 to 0 V, at a current density of 0.5 mA g−1. (b) Cycling performance and Coulombic efficiency of LVO-1 as an anode in a ARLB at a current density of 0.5 mAg−1 and (c) rate performance of LVO-1 for 10 cycles, each at current densities of 0.1 mA g−1, 0.5 mA g−1, 1.6 mA g−1, 3.2 mA g−1 and 6.4 mA g−1 in the cut-off voltage window −1.2 to 0 V vs. SCE. | ||
An increase in the aspect ratio of the LVO-1 rods leads to a higher electro-active surface area and reduced ion diffusion. This in turn, improves the mobility of lithium ions within the active material, enabling a larger number of lithium ions to intercalate into the active electrode material and occupy the vacant tetrahedral and octahedral sites in the crystal structure of LVO-1. The crystal structure, electrode porosity and composition of the materials also play very important roles in imparting high rate capabilities in anode materials.26 The pseudo-layered crystal structure of LVO-1 facilitates the lithium insertion–de-insertion into and from the layers via diffusion, while the presence of lithium ions (Li+) residing in the octahedral sites27–31 aids in maintaining the pseudo-layered structure, by supporting the V6O16− puckered layers, during the electrochemical lithiation–de-lithiation processes, resulting in an excellent electrochemical performance in terms of rate capability and cyclic stability.24,32 Good Coulombic efficiency in the range of 99–86% was achieved over cycles 1–100 at a high current density of 0.5 A g−1 (Fig. 3b).
After the electrochemical charge–discharge performance of LVO-1 as an anode, the electrode was examined again by XPS (Fig. 4). In the high resolution XPS spectrum of the V2p region, new peaks in both the V2p3/2 and V2p1/2 orbitals can be observed, along with the peaks corresponding to V5+ species. These new peaks can be attributed to V4+ species which can be believed to be generated during the charging process. Furthermore, the overall capacity of a material like LVO-1 can have different relative contributions of surface capacitive and diffusion controlled bulk intercalation processes, which can be measured using electrochemical methods.
 | ||
| Fig. 4 High-resolution XPS spectra of LVO-1 at the V2P region after charge–discharge, showing the existence of vanadium in the V5+/4+ oxidation states. | ||
Li3V6O16 has a pseudo-layered structure where the layers are held together by the two lithium ions present in the octahedral pockets of the structure. We differentiate the modes of charge storage mechanisms in Li3V6O16 nanowires, i.e. surface-capacitance and bulk lithium intercalation, by fitting the voltammetric currents at various sweep rates (Fig. 5a) to appropriate power law relationships given by eqn (3) derived from eqn (1) and (2) (see ESI†). Total charge storage depends exclusively on both capacitive and lithium intercalation processes. From Fig. 5b, the surface capacitance contribution of LVO-1 was computed to be ∼22.22% and the intercalation capacity was ∼77.83% at 4 mV s−1 while at 0.5 mV s−1 (Fig. S4†). Similarly, the surface capacitance contribution substantially decreased to ∼9.17% with a corresponding increase in the intercalation capacity to ∼90.82% (Fig. S4†). Usually surface-capacitance based energy storage behavior helps in stable operations at higher current densities (i.e. faster charge–discharge kinetics), thereby with better power density, while Li-ion intercalation based energy storage mechanisms help in higher capacity, and hence, increased energy density. Knowledge of the charge storage mechanism is necessary to tune the two Faradaic-based charge storage mechanisms, which play crucial roles in obtaining a battery with the desired functionality for different applications. Tuning the morphology, size of the material and replacement of the intercalating ion are some of the strategies that can be used to tune the charge storage mechanism.
 | ||
| Fig. 5 (a) Cyclic voltammetric response of LVO-1 rods at sweep rates of 4 mV s−1 (a), 2 mV s−1 (b), 1 mV s−1 (c), 0.5 mV s−1 (d) and 0.25 mV s−1 (e). (b) Charge storage mechanism using the inverse power law with the peak currents corresponding to the cycles (a–e), as shown in (a). | ||
Conclusions
In conclusion, here we have demonstrated the successful synthesis of LVO-1 having a rod-like morphology by a facile hydrothermal method, followed by subsequent heat treatment at 400 °C. Characterization via FE-SEM, TEM, and AFM revealed the formation of rod-like structures with a high aspect ratio of >20. X-ray diffraction confirmed the formation of 100% crystalline phase pure synthetic lithium vanadate bronze Li3V6O16 (LVO-1). The application of LVO-1 as an anode material in an aqueous lithium ion battery (ARLB) was demonstrated and the electrochemical properties were studied via cyclic voltammetry and galvanostatic charge–discharge cycling. A high initial capacity of >120 mA h g−1 at a high current rate was achieved. Upon cycling to 100 cycles, ∼52% of the initial capacity was retained, exhibiting the superior rate performance and cycling stability of LVO. More importantly, it can be deduced that the layered structure of LVO, coupled with the presence of three lithium ions, resulted in a stable structure, enabling the ease of lithium intercalation–deintercalation, suggesting future applications of LVO-1 as a potential electrode material for aqueous lithium ion batteries. The charge storage mechanism was studied by performing cyclic voltammetric experiments with LVO-1 as an anode at various voltage sweep rates, from which we observed a dominant bulk lithium ion intercalation based charge storage in LVO-1 compared to the intercalation and double-layer capacitance based charge storage.33 Further studies on LVO-1 could aim to improve its volumetric energy density by enhancing the diffusion controlled lithium insertion, along with the surface-capacitive charging, by tailoring different nanoarchitectures and alkali-metal ion replacement, which is ongoing.Acknowledgements
We acknowledge Timcal for providing Super P-Li Carbon black. These authors are grateful to The Nanyang Technological University and Ministry of Education, Singapore for financial support.Notes and references
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4270 CrossRef CAS.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243 CAS.
- Advanced in Lithium-ion Batteries, ed. W. van Schalkwijk and B. Scrosati, Kluwer Academic/Plenum, New York, 2002 Search PubMed.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4302 CrossRef CAS.
- Lithium Batteries Science and Technology, ed. G.-A. Nazri and G. Pistoia, Kluwer Academic/Plenum, Boston, 2004 Search PubMed.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- T. M. Bandhauer, S. Garimella and T. F. Fuller, J. Electrochem. Soc., 2011, 158, R1–R25 CrossRef CAS PubMed.
- G. L. Soloveichik, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 503–527 CrossRef CAS PubMed.
- Z. Yang, J. Zhang, M. C. W. Kinter-Meyer, X. Liu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- http://www.bbc.co.uk/news/business-21054089.
- http://www.technologyreview.com/news/521976/are-electric-vehicles-a-fire-hazard/.
- W. Li, J. R. Dahn and D. S. Wainwright, Science, 1994, 264, 1115–1118 CAS.
- W. Tang, Y. Zhu, Y. Hou, L. Liu, Y. Wu, K. P. Loh, H. Zhang and K. Zhu, Energy Environ. Sci., 2013, 6, 2093–2104 CAS.
- G. Wang, L. Fu, N. Zhao, L. Yang, Y. Wu and H. Wu, Angew. Chem., Int. Ed., 2006, 46, 295–297 CrossRef PubMed.
- H. Wang, K. Huang, Y. Zeng, S. Yang and L. Chen, Electrochim. Acta, 2007, 52, 3280–3285 CrossRef CAS PubMed.
- X.-H. Liu, T. Saito, T. Doi, S. Okada and J.-I. Yamaki, J. Power Sources, 2009, 189, 706–710 CrossRef CAS PubMed.
- H. Majunatha, G. S. Suresh and T. V. Venkatesha, J. Solid State Electrochem., 2011, 15, 431–445 CrossRef PubMed.
- J. Y. Luo, W. J. Cui, P. He and Y. Y. Xia, Nat. Chem., 2010, 2, 760–765 CrossRef CAS PubMed.
- J. Köhler, H. Makihara, H. Uegaito, H. Inoue and M. Toki, Electrochim. Acta, 2000, 46, 59–65 CrossRef.
- J. Jiang, Z. Wang and L. Chen, J. Phys. Chem. C, 2007, 111, 10707–10711 CAS.
- H. Liu, Y. Wang, L. Li, K. Wang, Y. Wang and H. Zhou, J. Power Sources, 2009, 192, 668–673 CrossRef CAS PubMed.
- H. Liu, Y. Wang, L. Li, K. Wang, E. Hosono and H. Zhou, J. Mater. Chem., 2009, 19, 7885–7891 RSC.
- N. A. Chernova, M. Roppolo, A. C. Dillon and M. S. Whittingham, J. Mater. Chem., 2009, 19, 2526–2552 RSC.
- G. Q. Liu, C. L. Zeng and K. Yang, Electrochim. Acta, 2002, 47, 3239–3243 CrossRef CAS.
- D. Zhou, S. Liu, H. Wang and G. Yan, J. Power Sources, 2013, 227, 111–117 CrossRef CAS PubMed.
- C. Fongy, A.-C. Gaillot, S. Jouanneau, D. Guyomard and B. Lestriez, J. Electrochem. Soc., 2010, 157, A885–A891 CrossRef CAS PubMed.
- J. Yu, J. C. Yu, W. Ho, L. Wu and X. Wang, J. Am. Chem. Soc., 2004, 126, 3422–3423 CrossRef CAS PubMed.
- W. Avansi, Jr, C. Ribeiro, E. R. Leite and V. R. Mastelaro, Mater. Chem. Phys., 2011, 127, 56–61 CrossRef PubMed.
- J. Kawakita, M. Majima, T. Miura and T. Kishi, J. Power Sources, 1997, 66, 135–139 CrossRef CAS.
- Y. Liu, X. Zhou and Y. Guo, Electrochim. Acta, 2009, 54, 3184–3190 CrossRef CAS PubMed.
- H. Wang, W. Wang, Y. Ren, K. Huang and S. Liu, J. Power Sources, 2012, 199, 263–269 CrossRef CAS PubMed.
- H. Wang, K. Huang, S. Liu, C. Huang, W. Wang and Y. Ren, J. Power Sources, 2011, 196, 788–792 CrossRef CAS PubMed.
- S. Liu, S. H. ye, C. Z. Li, G. L. Pan and X. P. Gao, J. Electrochem. Soc., 2011, 158, A1490–A1497 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra02804j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |