
Coordination variability of CuI in multidonor heterocyclic thioamides: synthesis, crystal structures, luminescent properties and ESI-mass studies of complexes†
Abstract
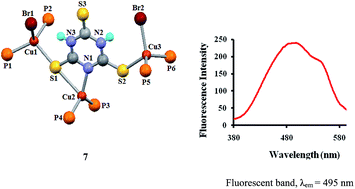
The chemistry of copper(I) with scarcely investigated heterocyclic thioamides, particularly 2,4,6-trimercaptotriazine, purine-6-thione, 2,4-dithiouracil, 2-thiouracil and pyrimidine-2-thione is described. The interaction of 2,4,6-trimercaptotriazine (tmtH3) with [Cu(CH3COO)(PPh3)2] resulted in a pair of bond isomers: [Cu(κ1N-tmtH2)(PPh3)2] (6a), [Cu(κ1N,κ1S-tmtH2)(PPh3)2] (6b); with copper(I) bromide and PPh3, it formed a trinuclear complex, [Cu3Br2(κ1N,κ1S, μ-S-tmtH2)(PPh3)6] (7) with anionic tmtH2−. The 2,4-dithiouracil with copper(I) halides (CuCl, CuBr) and PPh3 yielded dinuclear complexes: [Cu2(μ-Cl)(κ1S,κ1S-dtucH)(PPh3)4] (4) and [Cu2(μ-Br)(κ1S,κ1S-dtucH)(PPh3)4] (5) with unusual eight-membered metallacyclic rings. Pyrimidine-2-thione (pymSH) is an N,S-chelated anion in a mononuclear complex, [Cu(κ1N,κ1S-pymS)(PPh3)2] (1), whereas 2-thiouracil (tucH2) with copper(I) chloride and PPh3 yielded a tetrahedral complex, [CuCl(κ1S-tucH2)(PPh3)2] (3). Purine-6-thione (purSH2) coordinated to CuI in two different modes yielding mono- and di-nuclear complexes, [Cu(κ1N,κ1S-purS)(PPh3)2]·CH3OH (2a) and [Cu2(κ1N,μ-S-purS)2(PPh3)2] (2b). The existence of bond isomers (6a and 6b), synthesis of novel dinuclear (4 and 5) and rare trinuclear (7) complexes with unusual bonding patterns are the novel features of the present study. Complexes show intense emission bands in the 380–530 nm region with λem at 490 to 495 nm.
 Please wait while we load your content...
Please wait while we load your content...

