
Facile hydrothermal synthesis of a highly efficient solar active Pr6O11–ZnO photocatalyst and its multiple applications†
Abstract
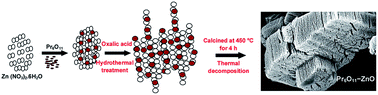
Coupled semiconductor oxide nanomaterial Pr6O11–ZnO was fabricated by a simple hydrothermal method and characterized by X-ray diffraction (XRD), field emission scanning electron microscopy (FE-SEM), elemental color mapping, high resolution transmission electron microscopy (HR-TEM), X-ray photoelectron spectroscopy (XPS), UV-vis diffuse reflectance spectroscopy (DRS), photoluminescence spectroscopy and BET surface area measurements. XRD analysis reveals that the as synthesized product has face-centered cubic phase of Pr6O11 and hexagonal wurtzite phase of ZnO. FE-SEM images of Pr6O11–ZnO show the nanochain like structures and Pr6O11 nanoparticles were homogeneously dispersed on the ZnO surface. The XPS analysis shows the presence of Zn, O and Pr elements and their oxidation states. Pr6O11–ZnO has increased absorption in the UV as well as visible region. The photocatalytic activity toward degradation of Acid Violet 7 (AV 7) under natural sunlight was investigated. Pr6O11–ZnO exhibits higher photocatalytic activity when compared to pure ZnO and Pr6O11 particles. Pr6O11–ZnO is more advantageous in toxic azodye degradation because of its reusability and higher efficiency at the neutral pH 7. Hydrophobicity of Pr6O11–ZnO has been evaluated using contact angle measurements. Pr6O11–ZnO modified TEOS coated substrates show significant hydrophobic properties. Pr6O11–ZnO exhibits high DC and photoconductivity, which make it useful for soliton wave communication and solar cell applications. Our results provide some new insights on the performance of a solar active photocatalysts, self cleaning and highly conductive materials.
 Please wait while we load your content...
Please wait while we load your content...

