
Study on intramolecular charge transfer processes, solvation dynamics and rotational relaxation of coumarin 490 in reverse micelles of cationic gemini surfactant†
Abstract
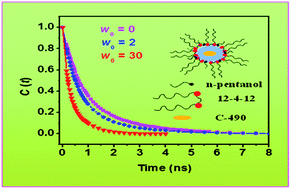
The steady-state fluorescence properties, steady-state fluorescence anisotropy, solvation dynamics and rotational relaxation of coumarin 490 (C-490) have been studied in cationic gemini surfactant 1,4-bis(dodecyl-N,N-dimethylammoniumbromide)butane (12-4-12)–cyclohexane–n-pentanol–water reverse micelles (RMs). C-490 molecule resides in the water pool of RMs. A lesser extent of hydrogen bonding interactions occurs between the –OH group of the alcohol and the C-490 molecule at the interface of the present RMs as compared to the RMs of reported conventional cationic surfactants. The effect of the more hydrophobic nature of coumarin 480 (C-480) used to study the RMs of conventional cationic surfactants cannot be ruled out. C-490 migrates to the water pool with increasing content of water molecules. Therefore, the results of steady-state fluorescence properties, rotational relaxation and solvation dynamics depend on the water loading of RMs, unlike the RMs of conventional cationic surfactants. Thus, the solvation dynamics in gemini surfactant RMs are one order of magnitude faster as compared to the RMs of conventional cationic surfactants. The faster solvation dynamics in gemini surfactant RMs as compared with the reported AOT RMs are due to the absence of hydrogen bonding interactions between the water molecules and quaternary ammonium headgroups of gemini surfactants. The polarity of the surrounding microenvironment is found to have very little effect on the rates of non-radiative processes in the water pool of gemini surfactant RMs. The rates of non-radiative processes in the present RMs are found to be higher than those in reported AOT RMs.
 Please wait while we load your content...
Please wait while we load your content...

