
Small band gap D-π-A-π-D benzothiadiazole derivatives with low-lying HOMO levels as potential donors for applications in organic photovoltaics: a combined experimental and theoretical investigation†
Abstract
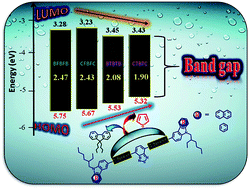
In an attempt to develop small organic molecules with potential applications as donors in organic photovoltaic (OPV) devices, we have synthesized and characterized four novel benzothiadiazole (A) core structured D-π-A-π-D dyes featuring carbazole and benzocarbazole as donors (D) and fluorene and thiophene as spacers (π). The effects of the π-spacer units and variations in donor strength on the photophysical, electrochemical and thermal properties of the molecules have been investigated in detail. The replacement of fluorene by thiophene as a π-spacer promotes planarity, resulting in a larger bathochromic absorption shift, enhanced emission profiles and an enhanced intramolecular charge transfer (ICT) transition. The introduction of the benzocarbazole unit creates a low-lying HOMO level, as inferred from cyclic voltammetry studies. All the dyes exhibit remarkable thermal robustness. Theoretical calculations have been carried out to understand the structure–property relationships of the synthesized materials. The results obtained from the characterization methods reveal that the dyes with thiophene π-spacers show better optoelectronic properties compared to their fluorene counterparts. Solution-processable bulk-heterojunction devices with a structure of ITO/PEDOT:PSS (38 nm)/active layer/Ca (20 nm)/Al (100 nm) were fabricated using the materials investigated in this study as donors and (6,6)-phenyl C61-butyric acid methyl ester (PC61BM) as an acceptor. A power conversion efficiency of 1.62% for the molecule with thiophene as a spacer and carbazole as donor/PC61BM was achieved for the preliminary photovoltaic devices under simulated AM 1.5 illumination (100 mW cm−2).
 Please wait while we load your content...
Please wait while we load your content...

