
Simple hydrothermal preparation of α-, β-, and γ-MnO2 and phase sensitivity in catalytic ozonation
Yuming Dong*,
Kun Li,
Pingping Jiang,
Guangli Wang,
Hongyan Miao,
Jingjing Zhang and
Chi Zhang
Key Laboratory of Food Colloids and Biotechnology (Ministry of Education of China), School of Chemical and Material Engineering, Jiangnan University, Wuxi 214122, P. R. China. E-mail: dongym@jiangnan.edu.cn; Fax: +86-510-85917763
First published on 13th August 2014
Abstract
The investigation of the roles of bonds and phases in catalysis is of great significance for designing novel catalysts and proposing mechanisms. Hindered by the lack of comparable catalyst samples, the influence of bonds and phases on the instantaneous reaction between ozone and MnO2 is still unknown. In this paper, α-, β-, and γ-MnO2 were prepared through a uniform hydrothermal method, and the phase sensitivity of the manganese dioxide catalysts in the ozonation process was investigated. Raman spectra indicated that bands transformed to a larger range in the window of 550–700 cm−1, which is considered to be the fingerprint region of manganese dioxides. During catalytic ozonation, ozone reacted with particular Mn–O bonds to generate more powerful oxygen species, leading to better oxidation efficiency compared to single ozonation. The active bonds, which are favourable to the activation of ozone, include bonds belonging to the pyrolusite type structure of γ-MnO2, corresponding to the [MnO6] octahedral frameworks of β-MnO2 and perpendicular to the direction of the [MnO6] octahedral double chains of α-MnO2. Thermogravimetric analysis confirmed that both active surface oxygen and lattice oxygen were responsible for the catalytic activity of α-MnO2, while lattice oxygen and the bonded manganese of MnO2 contributed to the catalytic activity of β- and γ-MnO2.
1. Introduction
Manganese dioxides (MnO2) are promising materials for many technological applications such as lithium batteries, oxidant agents, and catalysts.1–4 Their high activities are ascribed to their tunnel and layered structures, which have edge- or corner-sharing [MnO6] octahedra. Based on the law of varieties connectivity schemes, MnO2 shows great structural flexibility and appears in a number of crystallographic polymorphs such as α-, β-, and γ-MnO2.5 In addition, different connectivity schemes result in the anisotropism of Mn–O bonds, even in the same [MnO6] octahedron.6 Logically speaking, these bonds display differences in bond length, background energy state and so on. Nevertheless, depending on the nature of reactants, special Mn–O bonds or planes can be suitable for the absorption and activation of reactants. The investigation on the roles of bonds and phases in catalysis is of great significance for designing novel catalysts and proposing mechanisms.Catalytic ozonation is an attractive technology for organic pollutant degradation that does not require the control of temperature or pressure.7–11 Several articles12–15 have been published in the catalytic ozonation field using Mn-containing catalysts. Taking advantage of the particular properties of various catalysts, they revealed that certain interactions with reactants lead to a satisfactory consequence in pollutant degradation. The commonality of these catalysts was that manganese oxides played a vital role for catalysis. Some efforts have been made to identify the active site and oxygen species to determine the mechanism of catalytic ozonation. Limited by the varied preparation methods of the catalysts, a common essential factor in the catalytic activity could not be discerned.16 To date, the lack of comparable catalyst samples has limited the understanding of the influence of bonds and phases on the instantaneous reaction between ozone and MnO2. A uniform preparation strategy for different kinds of manganese dioxides is needed.
The objective of this paper is to investigate the relationship between the structure of manganese dioxide and catalytic ozonation activity. Using a facile hydrothermal method, α-, β- and γ-MnO2 were successfully obtained under similar conditions. They revealed different catalytic activities for the ozonation degradation of phenol. Control experiments were conducted to identify the phase sensitivity of MnO2 in the ozonation process. Thermogravimetric analysis was carried out to establish the correlation between surface active oxygen, lattice oxygen and catalytic activity.
2. Experimental
2.1 Materials
Potassium permanganate (KMnO4) and tetrahydrate manganese chloride (MnCl2·4H2O) were analytical grade and used without further purification. Deionized water was used throughout all experiments.2.2 Preparation of catalysts
The catalysts were synthesized by a simple hydrothermal method. In a typical procedure, a mixed solution (100 mL) containing MnCl2 and KMnO4 (total amount of 20 mmol) was stirred for 2 h and transferred into a Teflon-lined autoclave. The molar ratios of MnCl2 and KMnO4 are listed in Table 1. The autoclaves were then maintained at 160 °C for 6 h. The obtained brown precipitates were washed with water, dried at 80 °C and named Mn1, Mn2 and Mn3 (Table 1).| Sample | Raw materials ratio n(Mn2+)/n(MnO4−) | Phase | Lattice parameter (nm) | SBET(m2 g−1) | ||
|---|---|---|---|---|---|---|
| a | b | c | ||||
| a SEM images of catalysts: (a) Mn1, (b) Mn2 and (c) Mn3. | ||||||
| Mn1 | 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 :4 |
Pyrolusite: γ-MnO2 | 0.63279 | 1.04294 | 0.40319 | 2.53 |
| Mn2 | 7:3 |
Pyrolusite: β-MnO2 | 0.43953 | 0.43953 | 0.28542 | 2.93 |
| Mn3 | 5:5 |
Cryptomelane: α-MnO2 | 0.97401 | 0.98102 | 0.28427 | 28.78 |
2.3 Characterization of catalysts
The compositions and crystalline phases of the products were identified by powder X-ray diffraction (XRD) on a D8 X-ray diffractometer (Bruker AXS, German) using Cu Kα radiation (λ = 1.5406 Å). Scanning electron microscopy (SEM; S4800, Hitachi Co. Ltd., Japan) was employed to observe the morphologies and sizes. Thermogravimetric analysis (TGA) and differential thermal gravimetry (DTG) were performed with a Mettler TG/DTA b1/1100 SF analyser at a ramp rate of 10 °C min−1. Micro-Raman spectroscopy was conducted on an Invia-Reflex spectrometer (Renishaw). In a typical procedure, gases ozone was bubbled into water containing catalyst (1 g L−1, 25 °C). At specific time, a uniform thickness of sample was collected on a microscope slide. The specific surface areas of the catalyst samples were measured according to the Brunauer–Emmet–Teller (BET) method using nitrogen adsorption at 77.4 K on an ASAP 2020 instrument (Micromeritics, USA) after degassing under 120 °C for 2 h.2.4 Ozonation procedure and analytical methods
Catalytic activities were tested in the ozonation degradation of phenol. In each experiment, 200 mL of phenol aqueous solution (c0,phenol = 300 mg L−1) with a catalyst concentration of 1 g L−1 was used. After stirring for 10 min, 0.75 mg min−1 of ozone flow from a 3S-A5 laboratory ozoniser (Tonglin Technology, China) was bubbled into the above solution at 25 °C. Samples were collected every 15 min. Single ozonation (without any catalyst) was also carried out under the same conditions.The concentration of phenol was determined by high-performance liquid chromatography (HPLC, WATERS, USA) with a UV absorbance detector (UV 2487). The absorbance detection system for phenol was set at 210 nm. Elution was carried out by pumping methanol (HPLC/Spectro)–water (3:1 v/v) at a flow rate of 1.0 mL min−1. The ozone absorbance in aqueous solution was measured by a UV-vis spectrometer at 258 nm with a TU-1900 spectrophotometer (Beijing).
3. Results and discussion
3.1 Catalyst characterization
The XRD patterns of the as-synthesized samples are shown in Fig. 1. Depending on the ratio of raw materials, these manganese dioxide samples revealed obvious differences in crystalline phase, despite having the same synthetic procedures and conditions. With a raw material molar ratio [n(Mn2+)/n(MnO4−)] of 6:4, the product is γ-MnO2 (JCPDS 14-0644); several sharp diffraction peaks corresponding to the (120), (131) and (300) planes are observed. When the ratio is changed to 7:3, the crystalline phase is β-MnO2 (JCPDS 24-0735), with sharp peaks mainly corresponding to the (110), (101), (111), (211) and (312) planes. At a ratio of 5:5, the α-MnO2 crystalline phase (JCPDS 44-0141) with (110), (200), (310), (211), (301), (411) and (521) planes was obtained. Taking the intensities and half-widths of the diffraction peaks into account, the degree of the crystallinity follows the order: Mn2 > Mn3 > Mn1.
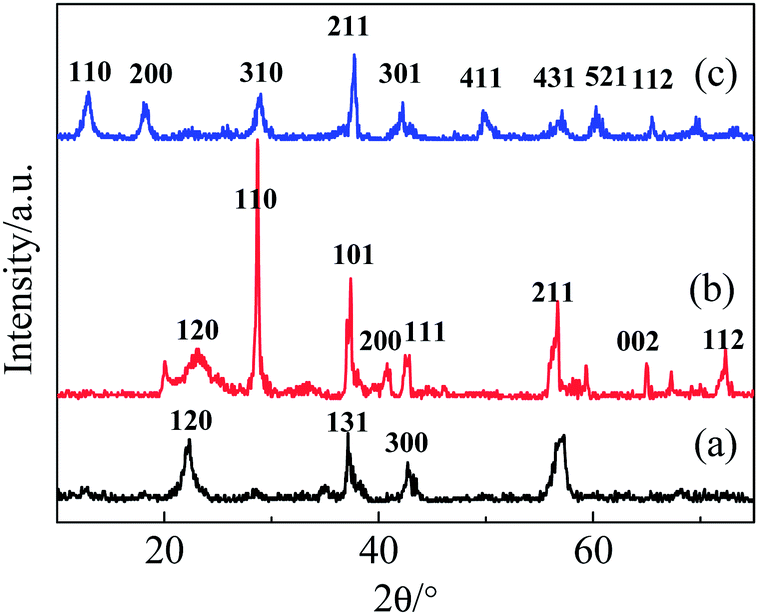 | ||
| Fig. 1 XRD patterns of (a) Mn1, (b) Mn2 and (c) Mn3. | ||
The lattice parameters calculated from the XRD patterns are listed in Table 1. From these lattice parameters, obvious differences among these samples can be observed. The structures and morphologies of samples were also substantially affected by the ratio of raw materials. As shown in Fig. 2, both Mn2 and Mn3 have one-dimensional rod morphologies with diameters and lengths in the range of 0.5–3.2 μm. Mn1 exhibits rods with dimensions of 3–5 μm, along with discrepant particles that wrap around the surface of the rods.
 | ||
| Fig. 2 SEM images of (a) Mn1, (b) Mn2 and (c) Mn3. | ||
As listed in Table 1, the BET surface areas of the samples are 2.53 (Mn1), 2.93 (Mn2), and 28.78 (Mn3) m2 g−1. In spite of their similar rod morphologies, the samples exhibit significant differences in surface area, which may be caused by their microstructures. The larger surface areas of Mn3 mostly resulted from the one-dimensional (2 × 2) tunnels in α-MnO2, which has a size of 0.46 nm × 0.46 nm (this will be discussed in detail in the Raman spectra analysis).17 By adjusting the [n(Mn2+)/n(MnO4−)] ratio in the simple hydrothermal method, α-, β- and γ-MnO2 were prepared under the same procedures and conditions. The α-, β-, and γ-MnO2 phases had 2 × 2 tunnels, 1 × 1 tunnels, and layer structures, respectively.18 The K+ ions can serve as templates in the formation of these tunnel or layer structures,19 although the tunnels may need more cations to be stable. Hence, α- and β-MnO2 phases were produced at high K+ ion concentrations, while the γ-MnO2 phase was obtained at a comparatively lower K+ ion concentration. Compared with α-MnO2, β-MnO2 has a large tunnel size that is suitable for the insertion/extraction of more K+ ions. These results are consistent with the literature.18,20–23 The preparation method used herein is easy to realize in a large-scale reactor, and it may provide a way to synthesize different kinds of MnO2 and further investigate the influence of phase.
3.2 Catalytic ozonation activity
As a probe action for catalytic ozonation activity, the degradation efficiency of phenol in presence of catalyst and ozone was investigated. The removal of phenol along with ozonation time is presented in Fig. 3. Generally, all the catalysts reveal high activity towards the oxidation of phenol. About 22% of phenol was removed in a single ozonation of 1 h, and 69% of phenol was removed in the presence of Mn3. Comparing the performance of these MnO2 catalysts, the trend of Mn3 > Mn1 ≈ Mn2 in terms of efficiency was observed. This observation indicates that the crystallography of MnO2 influenced the catalytic ozonation performance.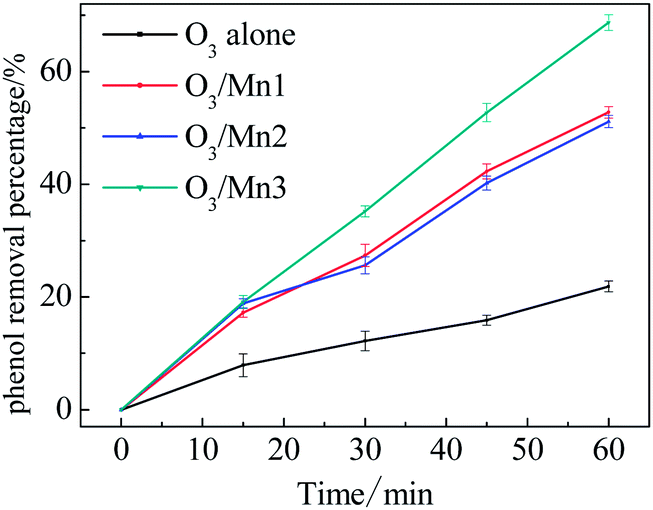 | ||
| Fig. 3 Evolution of phenol concentration of phenol in single ozonation and catalytic ozonation. | ||
Ozone decomposition experiments were then introduced to illustrate the interaction between catalyst and ozone. First, the catalyst was added into the O3-saturated solution. As shown in Fig. 4a, all the catalysts showed excellent activity towards the removal of ozone. Taking promotion percentage into account, the activities of these catalysts follow the order: Mn3 > Mn1 > Mn2. This sequence is consistent with the catalytic ozonation activity. Based on this result, it can be concluded that a strong interaction existed between ozone and catalyst.
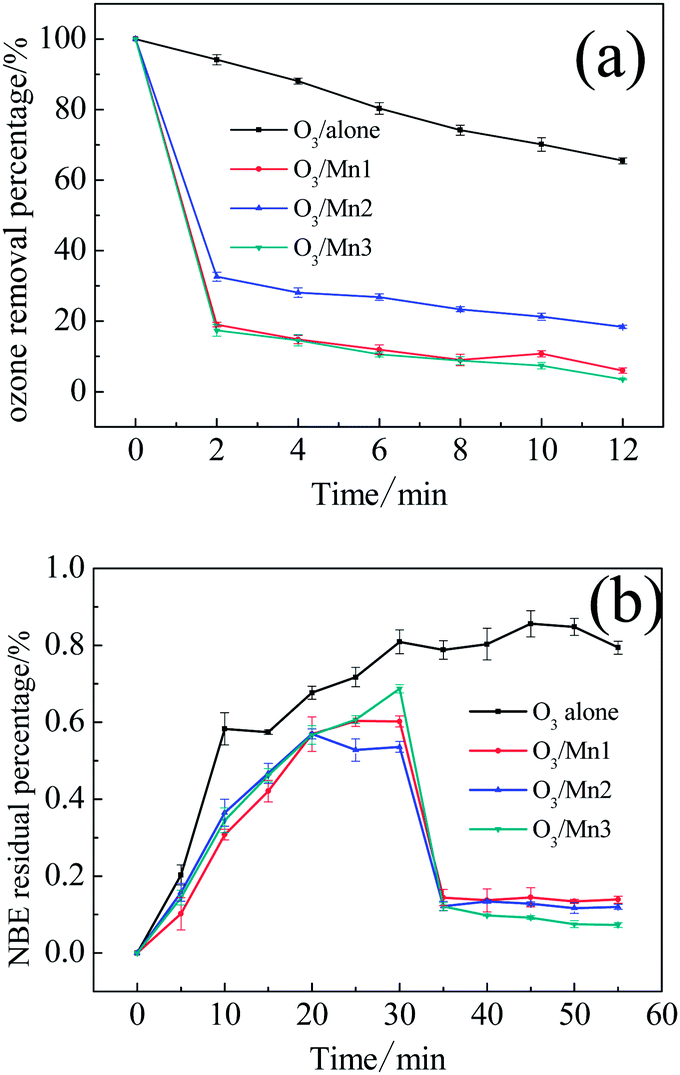 | ||
| Fig. 4 (a) Decomposition of ozone in O3-saturated aqueous dispersions of various catalysts; (b) ozone concentration in aqueous dispersions of various catalysts with the continued bubbling of ozone. | ||
To determine whether the removal of ozone was driven by absorption or decomposition, we continued bubbling gaseous ozone into water and added the catalysts after 30 min. As shown in Fig. 4b, absorbance of the O3-saturated aqueous solution without any catalyst remained at about 0.80 after 30 min. The addition of catalyst led to a sharp decline in ozone absorbance, and the absorbance value remained at a relatively low level in the following 20 min in presence of the three catalysts. The results demonstrate that the ozone removal resulted from catalytic decomposition.
3.3 Active surface atoms
The existence of surface active oxygen in MnO2 has been confirmed by XPS (X-ray photoelectron spectroscopy), TGA and other characterization methods.24,25 Several articles have also mentioned the importance of surface active oxygen, which plays a vital role in many reactions.26–28 Here, we employ TGA to detect the relationship between the catalytic activity and the surface and bulk oxygen atoms of these catalyst samples. Fig. 5 shows the TGA and DTG plots of each catalyst in the temperature range of 40–700 °C.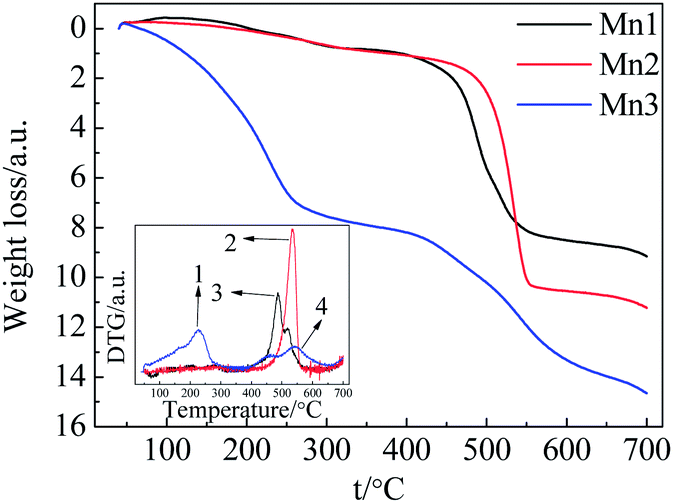 | ||
| Fig. 5 TGA and DTG plots of the catalysts under N2 atmosphere. | ||
For Mn1 and Mn2, the weight loss can be categorized into two segments. The initial weight loss before 300 °C was due to the removal of physically- and chemically-absorbed water. The subsequent weight loss from 400–560 °C was caused by the phase transformation from MnO2 to Mn2O3 (accompanied by the change in lattice oxygen and bonded manganese). The weight loss of 7.2% from 400–560 °C is smaller than the theoretical value of 9.2%, indicating the incomplete transformation from MnO2 to Mn2O3. In the case of Mn3, the weight loss includes three steps: the first step under 240 °C was caused by the removal of physically- and chemically-absorbed water; the second step from 240–300 °C possibly corresponded to the evolution of surface active oxygen; and the successive overlapping weight loss (total of 5.67%) from 400–640 °C originated from the transformation of MnO2 to Mn2O3 (accompanied by the change in lattice oxygen and bonded manganese).
In order to clarify which type of active atoms are responsible for ozonation catalysis, several control experiments were carried out. First, three catalyst samples were calcined at 600 °C for 3 h; these catalysts did not show any catalytic activity compared to the single ozonation (Fig. 6b). This indicated that all of the catalysts completely lost their activities after calcination. Second, the samples were calcined at 300 °C for 3 h. For Mn1 and Mn2, the intrinsic activities after calcination exhibited nearly no difference compared to the original catalysts, while a small decrease in activity was observed for Mn3 (Fig. 6a).
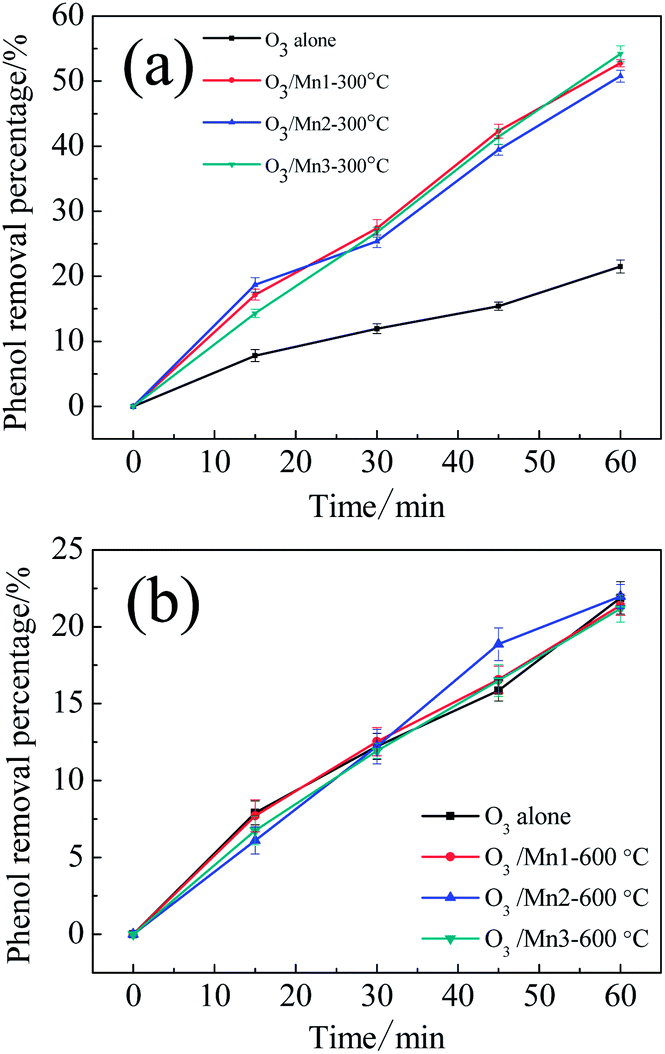 | ||
| Fig. 6 (a) Evolution of phenol concentration in single ozonation and catalytic ozonation with catalysts calcined at 300 °C; (b) evolution of phenol concentration in ozonation and catalytic ozonation with catalysts calcined at 600 °C. | ||
Considering the catalytic activity, weight loss analysis, and activity change after calcination for Mn1 and Mn2, it can be concluded that the lattice oxygen and the bonded manganese of MnO2 contributed to the catalytic activity these materials. In the case of Mn3, the notable phenomena are the partial loss of catalytic activity after calcination at 300 °C and the complete activity loss after calcination at 600 °C. Combined with the TGA results, the activity of Mn3 can be considered to have two origins: the lattice oxygen and bonded manganese of MnO2 and the active surface oxygen. With a surface active oxygen percentage of 1.86%, Mn3 exhibited a significantly different catalytic ozonation activity. The quantity of surface active oxygen had an obvious influence on the catalytic activity of Mn3.29 Consequently, active surface oxygen should play an important role in the activity of Mn3.
3.4 Raman spectra study
Almost all MnO2 structures can be described as a close-packed network of oxygen atoms in which Mn4+ cations are differently distributed to form [MnO6] octahedra (as shown in Fig. 7a). By sharing opposite octahedral edges or corners, the endless chains form and link to neighbouring octahedral chains. In this pattern, tunnel openings can be formed by basal layers.30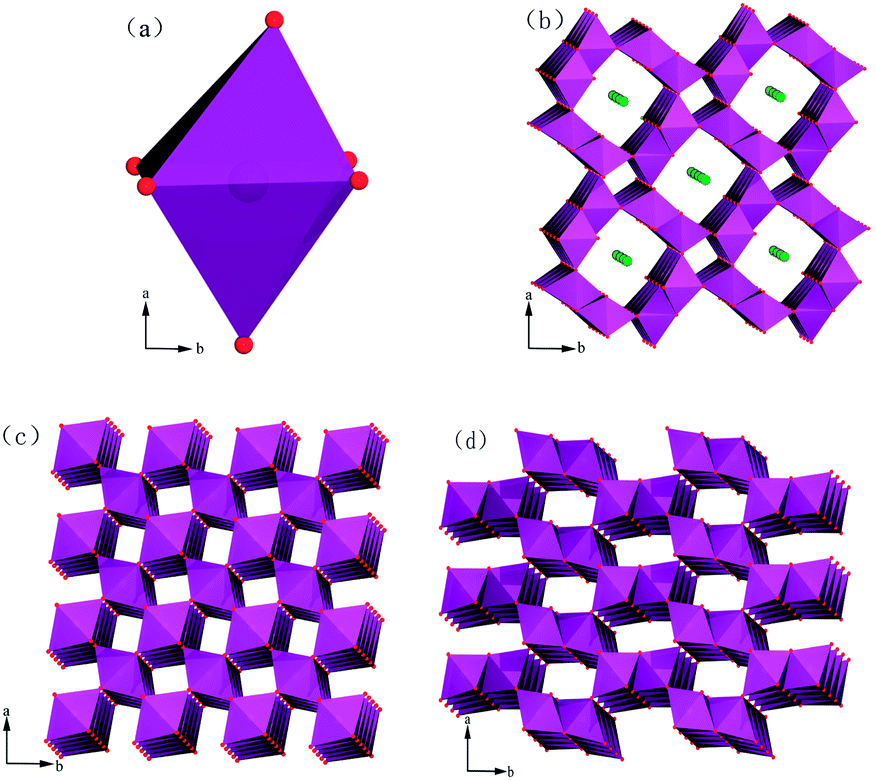 | ||
| Fig. 7 Polyhedral representations of the crystal structures of varieties of MnO2: (a) [MnO6] octahedron, (b) cryptomelane-type α-MnO2, (c) pyrolusite-type β-MnO2 and (d) pyrolusite-type γ-MnO2. | ||
The Raman spectra of these catalysts treated with ozone for different times are shown in Fig. 8. Compared with the two sharp bands situated at 550–700 cm−1 (this situation is suitable for these catalysts), other bands corresponding to the stretching mode of the Mn–O bond or skeletal vibrations of the [MnO6] chains are weak in intensity. In addition, the variations in Raman shift or peak intensity for these bands and peaks were negligible, even with a long period of ozone treatment. Thus, we only focus on the stronger peaks with significant changes and applied the same principle in the following analysis.
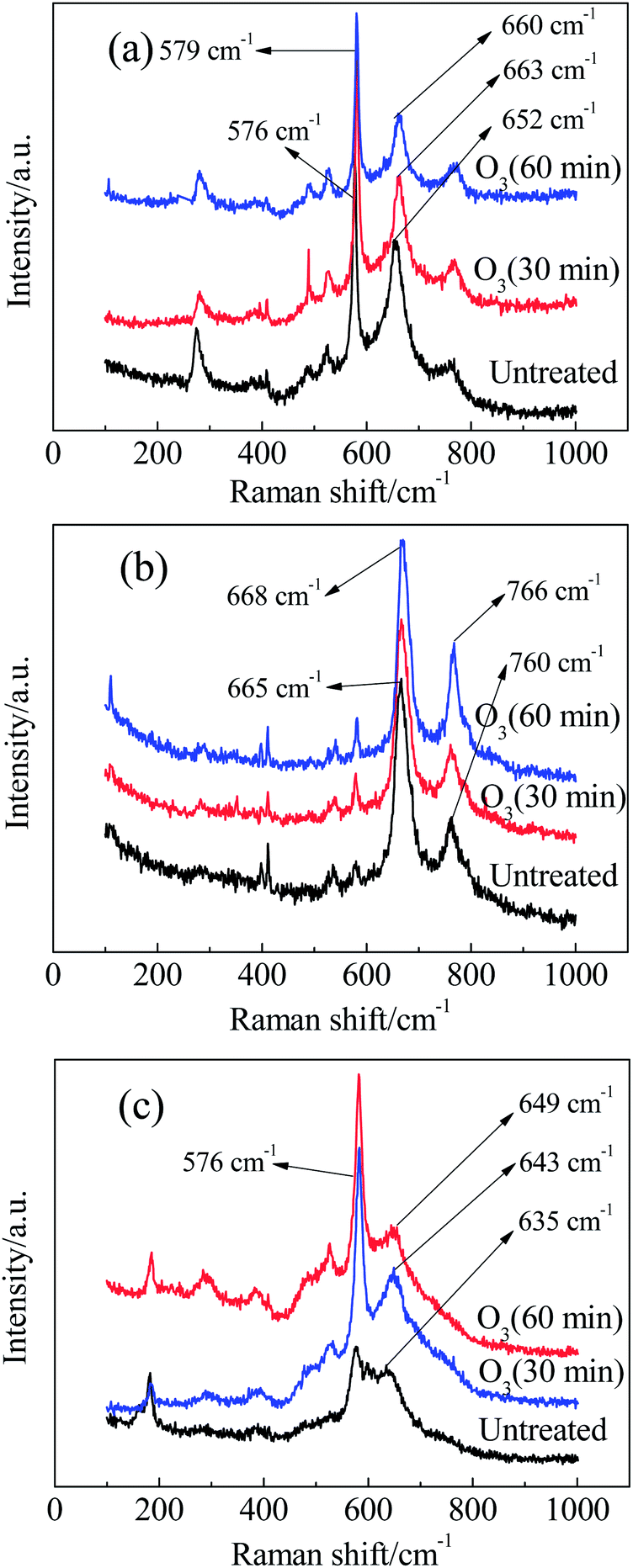 | ||
| Fig. 8 Raman spectra of (a) Mn1, (b) Mn2 and (c) Mn3 after ozone treatment for different times. | ||
First, we analyse the case of α-MnO2 (Mn3). Without any ozone treatment, two sharp diagnostic high-frequency Raman bands originating from the Ag spectroscopic species are situated at 635 and 576 cm−1 (Fig. 8c). On the basis of the principle for spectrum choice and analysis, 6Ag+6Bg+3Eg30–32 spectroscopic species are Raman active, and fifteen peaks or bands can be observed. However, some modes are difficult to observe due to the low polarisabilities and the overlap of incompletely resolved modes. According to previous studies, the above-mentioned two sharp peaks belong to the Mn–O stretching mode in [MnO6] octahedra. For cryptomelane-type α-MnO2, [MnO6] octahedra share edges with opposite octahedra to form an endless chain, which is in turn linked to neighbouring octahedral [MnO6] chains to form (2 × 2) or (1 × 1) tunnels (Fig.7b). The peak situated at 635 cm−1 might be related to Mn–O vibrations perpendicular to the direction of the [MnO6] octahedral double chains, and the Raman band at 579 cm−1 may correspond to the displacement of oxygen atoms relative to manganese atoms along the octahedral double chains.31
After treatment with ozone in water for 30 and 60 min, the peak at 635 cm−1 shifted to 643 and 649 cm−1, respectively. According to the quantum theory of Raman spectra, the interaction of a molecule with laser radiation can be analysed in terms of an energy-transfer mechanism. In other words, a change of state is accompanied by the gain or loss of one or more quanta of energy. Thus, Raman spectral features have been described in terms of frequency shifts, or, simply, the wave number relative to that of the excitation.33,34 For this reason, the Raman shift can measure the energy required for a bond to shift from the background state to the excited state. A larger Raman shift indicates a greater change in energy for the activation of bonds. In the presence of ozone, a little ozone energy may transfer to the Mn–O bond, resulting in the Raman shift of the Mn–O bond. Interestingly, the Raman shift returned to its original value when it was exposed to air for several hours. This indicates that the changes in Raman shift arising from ozone treatment are unstable, and the metastable state can recover to the original stable state by itself. This phenomenon is also observed in the other catalyst samples in this work. In conclusion, ozone reacted with the Mn–O bonds within [MnO6] octahedra perpendicular to the double chains to achieve a more unstable state by energy transfer from ozone to the Mn–O bond. As a result, ozone became a more powerful reactive oxidation species. In addition, ozone treatment changes the band shape and intensity of Mn3. The slope between the peaks at 635 cm−1 and 576 cm−1 became sharp after treatment with ozone, confirming the existence of active oxygen, which decreased the polarisability of the Mn–O bond. After ozone treatment, the average number of Mn–O bonds that can be irradiated by Raman laser increased, and the intensity of the Mn–O bonds then increased as well.35
Next, we discuss the case of β-MnO2 (Mn2). The Raman spectrum of Mn2 (Fig. 8b) displays two sharp bands: the band at 760 cm−1 is attributed to the B2g mode and involves antisymmetric Mn–O vibrations, while the more intense band at 665 cm−1 results from the A1g mode.36 This mode corresponds to the skeletal vibration of the Mn–O bond in the [MnO6] octahedral frame. The two bands shifted to 766 and 668 cm−1, respectively, when the ozone treatment time was extended to 60 min. This phenomenon implied that ozone acted preferentially on the Mn–O bonds in the [MnO6] octahedra.
Finally, we discuss the case of γ-MnO2 (Mn1). The [MnO6] octahedron chains formed (2 × 1) tunnels, as shown in Fig.7d.37 Mn1 is characterized by two sharp peaks at 652 and 576 cm−1 due to the stretching mode of the Mn–O bond in [MnO6] octahedra. It should be emphasized that the more intense band at 576 cm−1 belongs to ramsdellite γ-MnO2. It has been reported that a sharp band appears at 654 cm−1 when Pr (the rate of pyrolusite intergrowth) increased to 61%.37 Hence, the band at 652 cm−1 should correspond to pyrolusite intergrowth. When Mn1 was treated with ozone, some alternations also occurred. After treatment for 30 min, the band at 576 cm−1 did not change, while the peak at 652 cm−1 shifted to 663 cm−1. When the treatment time was extended to 60 min, two bands appeared at 660 and 579 cm−1. Therefore, pyrolusite γ-MnO2 reacts more easily with ozone than ramsdellite γ-MnO2. In other words, pyrolusite-type γ-MnO2 determined the catalytic activity of Mn1, while ramsdellite-type γ-MnO2 played a relatively small role.
4. Conclusion
α-, β- and γ-MnO2 have been prepared by a hydrothermal method. The preparation method is easy to realize in a large-scale reactor and may provide a way to synthesize different kinds of MnO2 and further investigate the influence of phase. Based on Raman shifts during ozonation, the sensitivity of the bonds to catalytic activity was determined. The observed active bonds are the Mn–O bonds perpendicular to the direction of the [MnO6] octahedral double chains of α-MnO2, the Mn–O bonds in the [MnO6] octahedral frameworks of β-MnO2 and the Mn–O bonds belonging to the pyrolusite-type structure of γ-MnO2. TGA results confirmed that active surface oxygen was positively correlated with the catalytic activity of α-MnO2, and that the lattice oxygen and bonded manganese of MnO2 contributed all the catalytic activity of β- and γ-MnO2.Acknowledgements
The authors gratefully acknowledge the support from the National Natural Science Foundation of China (no. 20903048, 21005031, 21275065), the National Key Technology Research and Development Program (2012BAD32B03-4), the Fundamental Research Funds for the Central Universities (JUSRP21113, JUSRP51314B), and MOE & SAFEA for the 111 Project (B13025).Notes and references
- X. Liu, Q. Wang, H. Zhao, L. Zhang, Y. Su and Y. Lv, Analyst, 2012, 137, 4552–4558 RSC.
- Y. Hasegawa, K. Fukumoto, T. Ishima, H. Yamamoto, M. Sano and T. Miyake, Appl. Catal., B, 2009, 89, 420–424 CrossRef CAS PubMed.
- H. C. Genuino, S. Dharmarathna, E. C. Njagi, M. C. Mei and S. L. Sui, J. Phys. Chem. C, 2012, 116, 12066–12078 CAS.
- E. E. McCabe and C. Greaves, Chem. Mater., 2006, 18, 5774–5781 CrossRef CAS.
- M. M. Thackeray, Prog. Solid State Chem., 1997, 25, 1–4 CrossRef CAS.
- C. M. Julien, M. Massot and C. Poinsignon, Spectrochim. Acta, Part A, 2004, 60, 689–700 CrossRef CAS.
- B. K. -Hordern, M. Ziółek and J. Nawrocki, Appl. Catal., B, 2003, 46, 639–669 CrossRef.
- J. Nawrocki and B. K. -Hordern, Appl. Catal., B, 2010, 99, 27–42 CrossRef CAS PubMed.
- L. Zhao, Z. Sun, J. Ma and H. Liu, Environ. Sci. Technol., 2009, 43, 2047–2053 CrossRef CAS.
- H. Yang, S. Yang, L. Wu and W. Liu, Catal. Commun., 2011, 15, 99–102 CrossRef CAS PubMed.
- J. Bing, L. Li, B. Lan, G. Liao, J. Zeng, Q. Zhang and X. Li, Appl. Catal., B, 2012, 115–116, 16–24 CrossRef CAS PubMed.
- S. Tong, W. Liu, W. Leng and Q. Zhang, Chemosphere, 2003, 50, 1359–1364 CrossRef CAS.
- Y. M. Dong, H. X. Yang, K. He, S. Q. Song and A. M. Zhang, Appl. Catal., B, 2009, 85, 155–161 CrossRef CAS PubMed.
- A. Lv, C. Hu, Y. Nie and J. Qu, Appl. Catal., B, 2010, 100, 62–67 CrossRef CAS PubMed.
- M. Sui, J. Liu and L. Sheng, Appl. Catal., B, 2011, 106, 195–203 CAS.
- M. Polverejan, J. C. Villegas and S. L. Suib, J. Am. Chem. Soc., 2004, 126, 7774–7775 CrossRef CAS PubMed.
- W. Hong, S. Iwamoto, S. Hosokawa, K. Wada, H. Kanai and M. Inoue, J. Catal., 2011, 277, 208–216 CrossRef CAS PubMed.
- X. Wang and Y. D. Li, Chem.–Eur. J., 2003, 9, 300–306 CrossRef CAS PubMed.
- Y.-F. Shen, S. L. Suib and C.-L. O'Young, J. Am. Chem. Soc., 1994, 116, 11020–11029 CrossRef CAS.
- S. Devaraj and N. Munichandraiah, J. Phys. Chem. C, 2008, 112, 4406–4417 CAS.
- Y. Khan, S. K. Durrani, M. Mehmood and M. R. Khan, J. Mater. Res., 2011, 26, 2268–2275 CrossRef CAS.
- W.-M. Chen, L. Qie, Q.-G. Shao, L.-X. Yuan, W.-X. Zhang and Y.-H. Huang, ACS Appl. Mater. Interfaces, 2012, 4, 3047–3053 CAS.
- D. Su, H.-J. Ahn and G. Wang, J. Mater. Chem. A, 2013, 1, 4845–4850 CAS.
- V. P. Santos, M. F. R. Pereira, J. J. M. Órfão and J. L. Figueiredo, Appl. Catal., B, 2010, 99, 353–363 CrossRef CAS PubMed.
- T. Rao, M. Q. Shen, L. W. Jia, J. J. Hao and J. Wang, Catal. Commun., 2007, 8, 1743–1747 CrossRef CAS PubMed.
- X. Tang, Y. Li, X. Huang, Y. Xu, H. Zhu, J. Wang and W. Shen, Appl. Catal., B, 2006, 62, 265–273 CrossRef CAS PubMed.
- H. Chen, A. Sayari, A. Adnot and F. Larachi, Appl. Catal., B, 2001, 32, 195–204 CrossRef CAS.
- M. A. Peluso, L. A. Gambaro, E. Pronsato, D. Gazzoli, H. J. Thomas and J. E. Sambeth, Catal. Today, 2008, 487, 133–135 Search PubMed.
- T. Gao, H. Fjellvåg and P. Norby, Anal. Chim. Acta, 2009, 648, 235–239 CrossRef CAS PubMed.
- T. Gao, M. Glerup, F. Krumeich, R. Nesper, H. Fjellvåg and P. Norby, J. Phys. Chem. C, 2008, 112, 13134–13140 CAS.
- C. M. Julien, M. Massot and C. Poinsignon, Spectrochim. Acta, Part A, 2004, 60, 689–700 CrossRef CAS.
- G. Thrrell and J. Corset, Raman Microscopy: Developments and Applications, 1996, pp. 2–5 Search PubMed.
- G. Gouadec and P. Colomban, Prog. Cryst. Growth Charact., 2007, 53, 1–56 CrossRef CAS PubMed.
- Q. Tu and C. Chang, Nanomedicine, 2012, 8, 545–558 CrossRef CAS PubMed.
- T. Gao, H. Fjellvåg and P. Norby, Nanotechnology, 2009, 20, 055610 CrossRef PubMed.
- Y. Xiong, Y. Xie, Z. Li and C. Wu, Chem.–Eur. J., 2003, 9, 1645–1651 CrossRef CAS PubMed.
- C. Julien, M. Massot, S. Rangan, M. Lemal and D. Guyomard, J. Raman Spectrosc., 2002, 33, 223–228 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |