DOI:
10.1039/C4RA02624A
(Paper)
RSC Adv., 2014,
4, 28439-28450
Synthesis and self-assembly behavior of organic–inorganic macrocyclic molecular brushes composed of macrocyclic oligomeric silsesquioxane and poly(N-isopropylacrylamide)
Received
25th March 2014
, Accepted 3rd June 2014
First published on 6th June 2014
Abstract
The novel organic–inorganic molecular brush composed of macrocyclic oligomeric silsesquioxane (MOSS) and poly(N-isopropylacrylamide) (PNIPAAm) (denoted by PNIPAAm@MOSS) was synthesized via the atom transfer radical polymerization (ATRP) approach. In bulk, the organic–inorganic molecular brushes were microphase-separated; the spherical MOSS microdomains with the diameter of 10–50 nm were dispersed into a continuous PNIPAAm matrix. Depending on the lengths of PNIPAAm chains, the PNIPAAm@MOSS molecular brushes were capable of self-assembling into cylindrical or spherical nano-objects in aqueous solutions as evidenced by transmission election microscopy (TEM) and dynamic light scattering (DLS). Both micro-differential scanning calorimetry (Micro-DSC) and ultraviolet-visible spectroscopy showed that the MOSS backbones exerted significant restriction of coil-to-globule transition of PNIPAAm chains.
Introduction
Responsive or “smart” materials have attracted considerable interest owing to their promising potential applications in biomedical fields.1–6 Poly(N-isopropylacrylamide) (PNIPAAm) is among the most frequently investigated thermoresponsive polymers. In aqueous solutions, PNIPAAm can display a lower critical solution temperature (LCST) of about 32 °C.7–11 Below this temperature, individual PNIPAAm chains adopt a random coil conformation, and the random coils collapse into globules upon heating the aqueous solutions up to about 32 °C; i.e., a so-called coil-to-globule transition occurs. It is realized that there are intermolecular hydrogen bonding interactions between amide groups of PNIPAAm and water molecular at lower temperatures, which promote the water solubility of this polymer. Upon heating the solution up to 32 °C or above, the intermolecular hydrogen bonding interactions are interrupted owing to the conformational changes in the PNIPAAm chains. In past decades, the thermoresponsive behavior of PNIPAAm was extensively investigated. These studies have revealed that some structural factors such as co-monomers, tacticity, crosslinking, grafting, molecular weights and end groups all affect the LCST behavior of PNIPAAm in aqueous solutions.12–18
Recently, considerable attention has been paid to the preparation of PNIPAAm amphiphiles containing hydrophobic structural units, because these PNIPAAm-containing amphiphiles are capable of self-assembling into a variety of thermoresponsive nano-objects in aqueous solutions. The self-assembly behavior of the PNIPAAm amphiphiles can exert a profound influence on the thermoresponsive behaviors of this polymer.8,19,20 For instance, Winnik et al. investigated the effect of the hydrophobic n-octadecyl termini on the merging of PNIPAAm mesoglobules and proposed that the rigidity and partial vitrification of mesoglobules may be the predominant cause of their stability against aggregation.8 Zhang et al.21 investigated the self-assembly of PS-b-PNIPAAm-b-PS triblock copolymers in aqueous solutions and reported the formation of flower-like aggregates with PNIPAAm interlocking rings and associated PS blocks as the core and PNIPAM rings as the coronas. It was found that the morphologies of micelle-like aggregates can significantly affect the collapse transition of PNIPAAm coronas. More recently, the PNIPAAm amphiphiles containing bulky and hydrophobic inorganic groups have attracted considerable interest owing to the specific architectures and self-assembly behavior of this class of organic–inorganic hybrids.22–28 Polyhedral oligomeric silsesquioxane (POSS)-capped semi-telechelic PNIPAAm hybrids have been synthesized via reversible addition-fragmentation chain transfer (RAFT) or atom transfer radical polymerization (ATRP);24,25 these organic–inorganic PNIPAAm amphiphiles can be self-assembled into thermoresponsive nano-objects. Wang et al.26 reported synthesis of α,ω-diPOSS-capped PNIPAAm telechelics with a “POSS-spreading” strategy. In this approach, a diPOSS-bearing trithiocarbonate was synthesized and used as the chain transfer agent, with which the RAFT polymerization of NIPAAm was carried out and two ends of PNIPAAm chain were capped with hepta(3,3,3-trifluropropyl) POSS. Owing to the high hydrophobicity of the POSS end groups, these POSS-capped PNIPAAm telechelics can form organic–inorganic physical hydrogels in water. These physical hydrogels displayed fast deswelling and reswelling properties compared with traditional chemically crosslinked hydrogels. Kuo et al.27 reported the synthesis of octa-armed PNIPAAm stars with POSS cores. It was found that these organic–inorganic PNIPAAm stars undergo a sharp coil-to-globule transition in water at about 32 °C, which is similar to linear PNIPAAm homopolymers. More recently, He et al.28 investigated the thermoresponsive and pH dual-responsive behaviors of a star-like block copolymer with a POSS core and poly(N-isopropylacrylamide)-block-poly(acrylic acid) diblock copolymer arms. The results showed that with the star-block architecture, the high local chain density near the POSS core may lead to enhanced intramolecular interactions between different types of units and hence significantly alter the critical phase transition behavior.
Polyhedral oligoalkylmetallasiloxanes are a class of novel organometallic compounds that consist of stereoregular alkylsiloxane macrocycles coordinated to alkaline and/or transition metals (e.g., Mn, Co, Ni, Cu and trivalent lanthanide metals).29–37 These metallasiloxane coordinates can conveniently be converted into macrocyclic oligomeric silsesquioxanes (MOSS) with satisfactory yields, which have potential for use as a new family of building blocks for organic–inorganic hybrids.38–41 In the present work, we explored the synthesis of the organic–inorganic macrocyclic molecular brushes composed of a MOSS backbone and PNIPAAm arms. Toward this end, we first synthesized a twenty-four membered MOSS macromer bearing twelve 2-chloropropionate moieties; the functionalized MOSS was then used as a macroinitiator for the atom transfer radical polymerization of N-isopropylacrylamide (NIPAAm) to afford the organic–inorganic macrocyclic molecular brushes. The purpose of this work is twofold: (i) to explore the synthesis of organic–inorganic macrocyclic molecular brushes with an inorganic macrocyclic molecule as the backbone and PNIPAAm as side chains; and (ii) to investigate the effect of organic–inorganic macrocyclic brush architectures on the thermoresponsive and self-assembly behavior of PNIPAAm. In the past years, the thermoresponsive and self-assembly behavior of a variety PNIPAAm amphiphiles has been extensively investigated. To the best of our knowledge, nonetheless, there has been no previous report on organic–inorganic PNIPAAm macrocyclic brushes. In this work, the thermoresponsive and self-assembly behavior of organic–inorganic macrocyclic molecular brushes were addressed on the basis of the results of transmission electronic microscopy (TEM), micro-differential scanning calorimetry (Micro-DSC), UV-vis spectroscopy and dynamic laser scattering (DLS).
Experimental
Materials
Organic silanes (i.e., methyltriethoxysilane and dimethylchlorosilane) were purchased from Gelest Co, USA and used as received. Copper(II) chloride (CuCl2) was obtained from Ruanshi Chemical Co, Jiangsu, China. Sodium hydroxide (NaOH), silver nitrate (AgNO3), pyridine and organic solvents such as anhydrous ethanol, methanol and toluene were purchased from Shanghai Reagent Co, Shanghai, China. Before use, toluene and pyridine were distilled over calcium hydride (CaH2) and then stored in a sealed vessel in the presence of a 4 Å molecular sieve. N-Isopropylacrylamide (NIPAAm) was prepared in this lab via the reaction between isopropylamine and acryloyl chloride. Copper(I) chloride (CuCl) was of chemically pure grade, supplied by Shanghai Reagent Co. China. Tris(2-(dimethylamino)ethyl)amine (Me6TREN) was prepared by following the methods reported by Matyjaszewski et al.42 All other solvents used were obtained from commercial sources. Before use, tetrahydrofuran (THF) was refluxed above sodium and distilled; triethylamine (TEA) and isopropyl alcohol (IPA) were dried over CaH2 and then distilled; N,N-dimethylformamide (DMF) was dried over anhydrous magnesium sulfate and distilled under reduced pressure.
Synthesis of dodecamethyldodeca(dimethylhydro)cyclododecasilsesquioxane (1)
First, the coordinate of the metals (i.e., alkaline and copper) with methylsiloxanolates was prepared via the reactions between methyltriethoxysilane [MeSi(OEt)3] and copper(II) chloride (CuCl2) in a basic media (e.g., NaOH) by following the methods reported by Shchegolikhina et al.43,44 Second, the coordinate (8.480 g, 4.99 mol) was added to the mixture composed of anhydrous toluene (160 mL), dimethylchlorosilane [(CH3)2SiHCl] (42.000 g, 442.1 mmol) and pyridine (28.400 g, 359.1 mmol) at room temperature. The reaction was carried out at 25 °C for 24 hours with vigorous stirring. The mixture was filtered to isolate the precipitates, and the solution was washed with deionized water until no chlorine ions were detected using an aqueous solution of silver nitrate (AgNO3). After drying with anhydrous MgSO4, all the solvents were eliminated with rotary evaporation to afford 7.36 g viscous liquid (i.e., compound 1) with the yield of 98.0%. 1H NMR (CDCl3): 4.72 (m, 1H, SiH), 0.205–0.194 [d, 6H, Si(CH3)2], 0.10 (s, 3H, O3SiCH3). 29Si NMR (ppm, CDCl3): −66.2 (d, SiO3/2), −6.4 [s, Si(CH3)2H].
Synthesis of allyl 2-chloropropionate (2)
To a flask equipped with a magnetic stirrer, dichloromethane (80 mL), triethylamine (18.200 g, 180 mmol) and allyl alcohol (8.350 g, 144 mmol) were charged with vigorous stirring and then 2-chloropropionyl chloride (18.0 g, 141.7 mmol) was added at 0 °C. The reaction was performed at 0 °C for 2 hours and at room temperature for 24 hours. The mixture was washed with deionized water, and the solvent was eliminated via rotary evaporation. The product (19.400 g) was obtained via distillation at reduced pressure with the yield of 92.1%. 1H NMR (ppm, CDCl3): 5.96–5.87 (m, 1H, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2OOCCHClCH3), 5.38–5.26 (m, 2H, CH2 = CHCH2OOCCHClCH3), 4.68–4.66 (m, 2H, CH2CHCH2OOCCHClCH3), 4.45–4.39 (q, 1H, CH2CHCH2OOCCHClCH3), 1.71–1.69 (d, 3H, CH2CHCH2OOCCHClCH3).
CHCH2OOCCHClCH3), 5.38–5.26 (m, 2H, CH2 = CHCH2OOCCHClCH3), 4.68–4.66 (m, 2H, CH2CHCH2OOCCHClCH3), 4.45–4.39 (q, 1H, CH2CHCH2OOCCHClCH3), 1.71–1.69 (d, 3H, CH2CHCH2OOCCHClCH3).
Synthesis of MOSS macromolecular initiator (3)
To a flask equipped with a dried magnetic stirrer, anhydrous toluene (24 mL), compound 1 [i.e., dodecamethyldodeca(dimethylhydro)cyclododecasilsesquioxane] (2.400 g, 1.492 mmol) and compound 2 (viz. allyl 2-chloropropionate) (4.000 g, 26.88 mmol) were charged with vigorous stirring. The flask was connected onto a Schlenk line to degas via three freeze–pump–thaw cycles, and Karstedt catalyst (80 μL) was added with a syringe. The reaction was carried out at 85 °C for 10 hours. The solvent and unreacted allyl 2-chloropropionate were eliminated via rotary evaporation, and the product (4.600 g) (i.e., compound 3) was obtained with the yield of 92.0%. 1H NMR (ppm, CDCl3): 4.42–4.30 (q, 1H, CHCl), 4.16–4.06 (s, 2H, COOCH2), 1.75–1.61 (d, 5H, ClCHCH3, CH2CH2Si), 0.63–0.54 [t, 2H, CH2CH2Si], 0.20–0.03 [t, 9H, O3SiCH3, O2Si(CH3)2]. 29Si NMR (ppm, CDCl3): −66.2 (d, SiO3/2), −6.4 [s, Si(CH3)2CH2CH2CH2OOCCHClCH3].
Synthesis of organic–inorganic molecular brushes
Typically, to a flask equipped with a magnetic stirrer, the above MOSS macromolecular initiator (i.e., compound 3) (0.18 g, 0.64 mmol with respect of allyl 2-chloropropionate group), NIPAAm (2.70 g, 23.8 mmol) and isopropyl alcohol (6 mL) were charged with vigorous stirring. The flask was connected onto the Schlenk line to degas via three freeze–pump–thaw cycles, and then copper(I) chloride (29.60 mg) and Me6Tren (87 μL) were added. The system was purged with highly pure nitrogen for 45 min and the polymerization was performed at room temperature for 24 hours. After that, 20 mL of tetrahydrofuran was added to dissolve the reacted product, and the solution was passed through a neutral alumina column to remove the catalyst. The solution was concentrated via rotary evaporation and then dropped into 200 mL of petroleum ether to afford the precipitates. The precipitates were re-dissolved in tetrahydrofuran and the as-obtained solution was re-dropped into petroleum ether to obtain the precipitates. After dried in vacuo at 30 °C for 24 hours, the polymer (2.1 g) was obtained with the conversion of PNIPAAm to be 71%. 1H NMR (400 MHz, CDCl3): 6.06–6.57 [m, –NHCH(CH3)2], 4.02 [s, NHCH(CH3)2], 2.4–1.2 [m, CH2CH], 1.14 (d, CH(CH3)2), 0.02–0.18 [m, O3SiCH3 and O2Si(CH3)2]. GPC: Mn = 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 with Mw/Mn = 1.46.
200 with Mw/Mn = 1.46.
Measurement and techniques
Nuclear magnetic resonance spectroscopy (NMR). The 1H NMR measurement was carried out on a Varian Mercury Plus 400 MHz NMR spectrometer at 25 °C and the 29Si NMR spectra were obtained on a Bruker Avance III 400 MHz NMR spectrometer. The samples were dissolved with deuterated chloroform (CDCl3) and the solutions were measured with tetramethylsilane (TMS) as an internal reference.
Matrix-assisted ultraviolet laser desorption/ionization time-of-flight mass spectroscopy (MALDI-TOF-MS). Gentisic acid (2,5-dihydroxybenzoic acid, DHB) was used as the matrix with dichloromethane as the solvent. The MALDI-TOF-MS experiment was carried out on an IonSpec HiResMALDI mass spectrometer equipped with a pulsed nitrogen laser (λ = 337 nm; pulse with = 3 ns). This instrument operated at an accelerating potential of 20 kV in reflector mode. Sodium was used as the cationizing agent; all data shown are for positive ions.
Gel permeation chromatography (GPC). The molecular weights and molecular weight distribution of polymers were determined on a Waters 717 Plus autosampler gel permeation chromatography apparatus equipped with Waters RH columns and a Dawn Eos (Wyatt Technology) multiangle laser light scattering detector; the measurements were carried out at 25 °C with dimethylformamide (DMF) as the eluent, at the rate of 1.0 mL min−1.
Micro-differential scanning calorimetry (Micro-DSC). The Micro-DSC measurements were performed with a SETARAM micro-DSC III system under a dry nitrogen atmosphere and circulating water. Before the measurement, the specimens were maintained at 18 °C for 15 min and were then heated to 60 °C at the heating rate of 1 °C min−1. The temperature at the minimum of the exothermic peak was taken as the lower critical solution temperature (LCST).
Determination of cloud point. Each polymer (0.01 g) was dissolved in 0.5 mL tetrahydrofuran (THF) and then the solution was added dropwise to 50 mL ultrapure water by a dropping funnel with vigorous stirring. After that, the suspension was stirred for additional 30 min, and a transparent emulsion was formed with the concentration of the polymer being 0.2 g L−1. The solvent (i.e., THF) in the suspension was eliminated via rotary evaporation for 2 hours. The cloud points were determined with an Agilent Technologies Cary 60 Ultraviolet-visible spectrometer at the wavelength of light λ = 550 nm. In the measurements, a thermostatically controlled cuvette holder was employed and the plots of light transmittance as a function of temperature were obtained.
Dynamic laser scattering (DLS). The above suspensions of PNIPAAm@MOSS at the concentration of 0.2 g L−1 were subjected to dynamic laser scattering (DLS). The laser light scattering experiments were conducted on a Malvern Nano ZS90 equipped with a He–Ne laser operated at the wavelength of λ = 633 nm; the data were collected at a fixed scattering angle of 90°.
Transmission electron microscopy (TEM). The morphological observations were conducted on a JEOL JEM 2100F electron microscope at a voltage of 200 kV. To investigate the morphology of the samples in bulk, the THF solutions of the polymers at the concentration of 1 wt% were cast on carbon-coated copper grids, and the solvent was slowly evaporated at room temperature and then in vacuo at 30 °C for 2 hours. The specimens were directly observed without an additional staining step. To investigate the self-assembly behavior of the samples in aqueous solutions, the specimens were prepared by dropping the suspensions of the polymer in water (about 10 μL at 0.2 g × L−1) onto carbon-coated copper grids; the water was eliminated via freeze drying.
Results and discussion
Synthesis of organic–inorganic molecular brushes
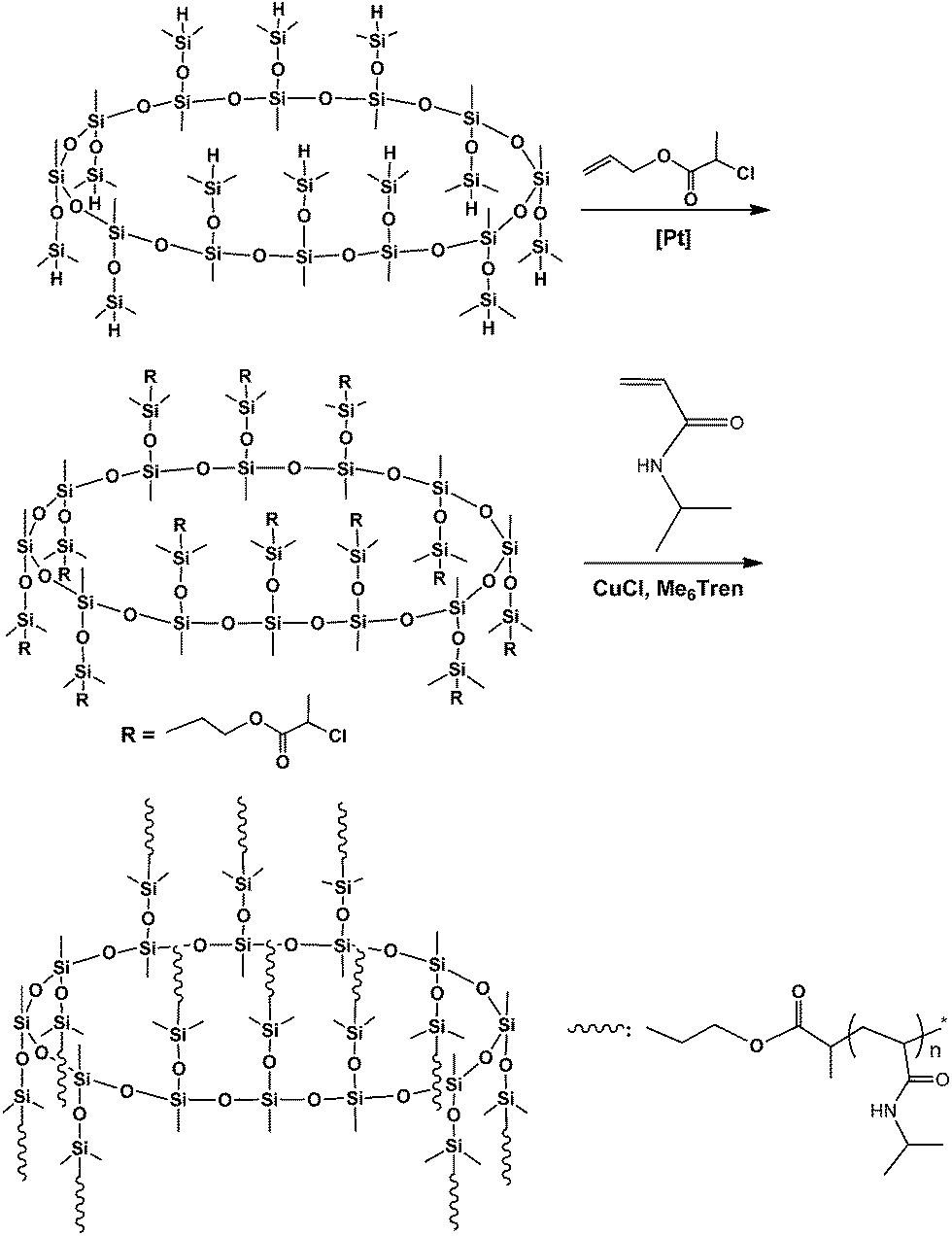
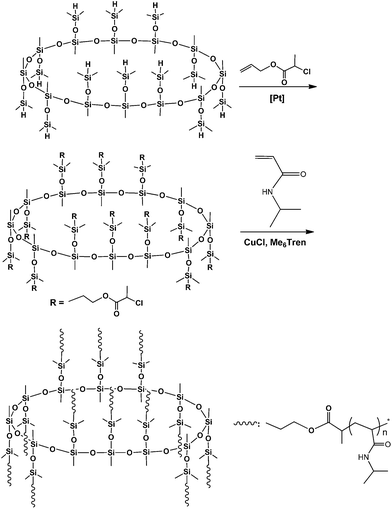
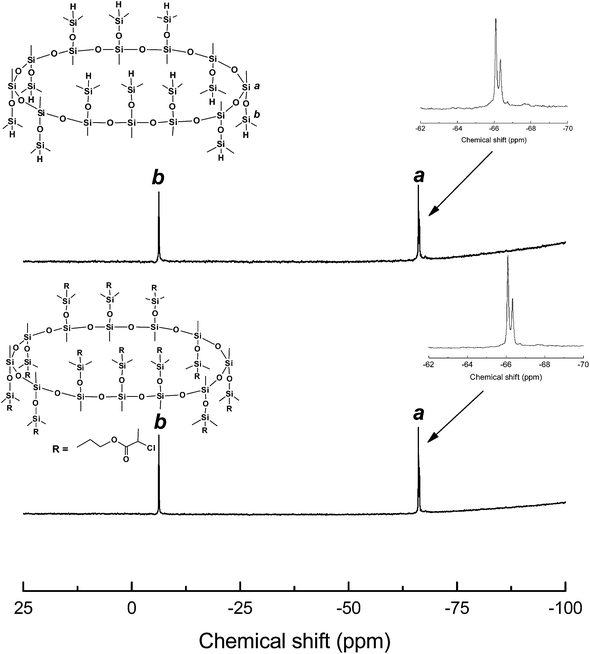
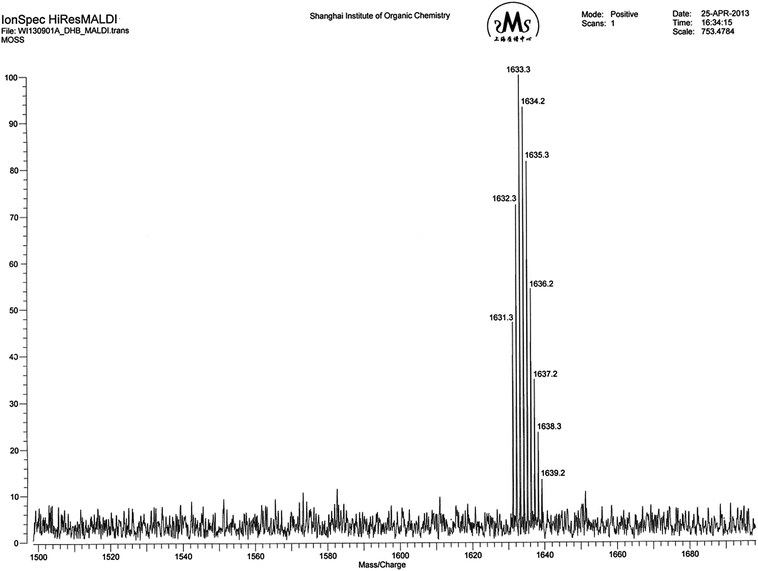
The route of synthesis for the organic–inorganic macromolecular brushes composed of macrocyclic oligomeric silsesquioxane (MOSS) and poly(N-isopropylacrylamide) (PNIPAAm) (denoted by PNIPAAm@MOSS) is shown in Scheme 1. First, the MOSS macromolecular initiator bearing 2-chloropropionate moieties was synthesized via the hydrosilylation reactions of dodecamethyldodeca (dimethylhydro)cyclododecasilsesquioxane (compound 1) with allyl 2-chloropropionate (compound 2). Compound 1 was synthesized via the silylation reaction of the coordinate of copper and sodium with methysiloxanolate with dimethylchlorosilane. The metallasilsesquioxanes used in this work were prepared via the reaction of methyltriethoxylsiloxane [CH3Si(OCH2CH)3] with sodium hydroxide (NaOH) and copper(II) chloride (CuCl2) in the presence of water and alcohol by following the method reported by Sergienko et al.43,44 It was reported that the crystal of the coordinate possessed a globular structure, in which the twelve-SiO3/2-membered organosiloxanolate ligand [i.e., vinylSi(O)O−1]12 in the tris-cis-tris-trans configuration constituted a saddle conformation, which was fixed by four Cu(II) ions. The metallacyclosiloxanolate was allowed to react with the dimethylchlorosilane to afford the macrocyclic oligomeric silsesquioxane bearing twelve Si–H bonds (compound 1). The 29Si NMR spectrum of compound 1 is shown in Fig. 1. In the 29Si NMR spectrum, two resonance peaks were detected at −6.4 and −66.2 ppm, respectively. The former is assignable to the silicon nucleus bonded with hydrogen atom (i.e., that in the Si–H bond), whereas the latter is assigned to the nucleus of silicon of macrocyclic backbones. The appearance of the two resonance peaks indicates that the compound had a macrocyclic structures with silsesquioxane as the backbone and hydrodimethylsiloxyl as side groups. It should be pointed out that the baseline uplifting of the spectral line in the range of −75 to −100 ppm resulted from the resonance of 29Si nucleus in the NMR tube. The expanded 29Si NMR spectrum shows that the signal of resonance at −66.2 ppm was composed of two splitting peaks with very close chemical shifts; the ratio of integral intensity of the peak at −66.1 ppm to that at −66.4 ppm was 2:1. The splitting of silicon resonance signal was related to the configuration of the MOSS macromer. It is proposed that trimethylsiloxyl groups appeared in the 24-membered macrocyclic silsesquioxane with the tris-cis-tris-trans configuration. Compound 1 was subjected to MALDI-TOF mass spectroscopy to measure its molecular weight; its mass spectrum is shown in Fig. 2. A group of intense peaks centered at the value of m/z equal to 1633.3 were detected, implying that the compound possessed the molecular weight of 1610 Da. The molecular weight is identical to the value estimated according to the formula of compound 1. The 29Si NMR and MALD-TOF mass spectroscopy results indicate that the MOSS macromer bearing twelve Si–H bonds was successfully obtained.
 |
| | Scheme 1 Synthesis of organic–inorganic molecular brushes composed of MOSS and PNIPAAm. | |
 |
| | Fig. 1 29Si NMR spectrum of dodecamethyldodeca(dimethylhydro) cyclododecasilsesquioxane (1). | |
 |
| | Fig. 2 MALDI-TOF mass spectrum of dodecamethyldodeca(dimethylhydro) cyclododecasilsesquioxane (1). | |
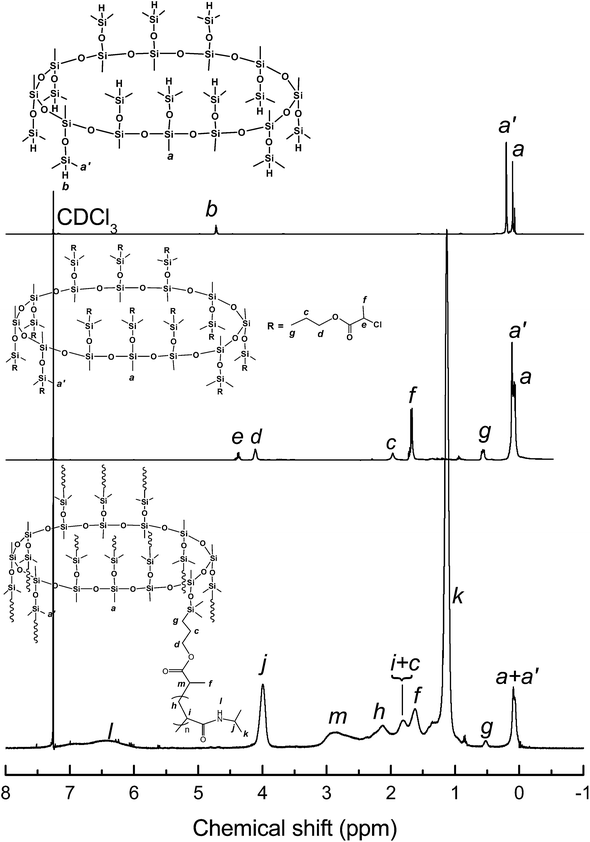
Hydrosilylation reaction of the MOSS macromer (viz. compound 1) with allyl 2-chloropropionate (2) was carried out to afford dodecamethyldodeca(dimethylpropyl-2-chloropropionate)cyclododecasilsesquioxane (compound 3), i.e., a MOSS macromolecular initiator bearing 2-chloropropionate moieties. Also shown in Fig. 1 is the 29Si NMR spectrum of compound 3. Compared with the 29Si NMR spectrum of compound 1, the positions of the resonance peaks assignable to the silicon nucleus of MOSS backbone (at −66.2 ppm) and the side groups (at −6.4 ppm) remained almost invariant, indicating that the hydrosilylation reaction did not alter the structure of the macrocyclic silsesquioxane. Shown in Fig. 3 are the 1H NMR spectra of compounds 1 and 3. For compound 1, the resonance peaks of methyl and Si–H protons were detected at 0.09, 0.203 and 4.73 ppm, respectively. The former two peaks are assignable to the protons of methyl groups connected to silsesquioxane backbone and the methyl in dimethylhydrosiloxayl groups, respectively. The resonance at 4.73 ppm is attributable to the protons in Si–H bonds. The ratio of integral intensity for these protons was measured to be 3:6:1, which is in good agreement with the value calculated according to the structural formula of compound 1. Compared with the 1H NMR spectrum of compound 1, the signal of resonance at 4.73 ppm assignable to the protons of Si–H bonds completely disappeared in the 1H NMR spectrum of compound 3, suggesting that the hydrosilylation has been performed to completion. Concurrently, several new signals of resonance appeared at 0.57, 1.69, 2.45, 1.98, 4.11 and 4.38 ppm, which are assignable to the protons of propyl 2-chloropropionate moiety as indicated in this figure. The 1H and 29Si NMR spectroscopy results indicate that the MOSS macromer bearing 2-chloropropionate moiety (compound 3) was successfully obtained.
 |
| | Fig. 3 1H NMR spectra of dodecamethyldodeca(dimethylhydro) cyclododecasilsesquioxane (1), MOSS macromolecular initiator (3) and PNIPAAm2K@MOSS. | |
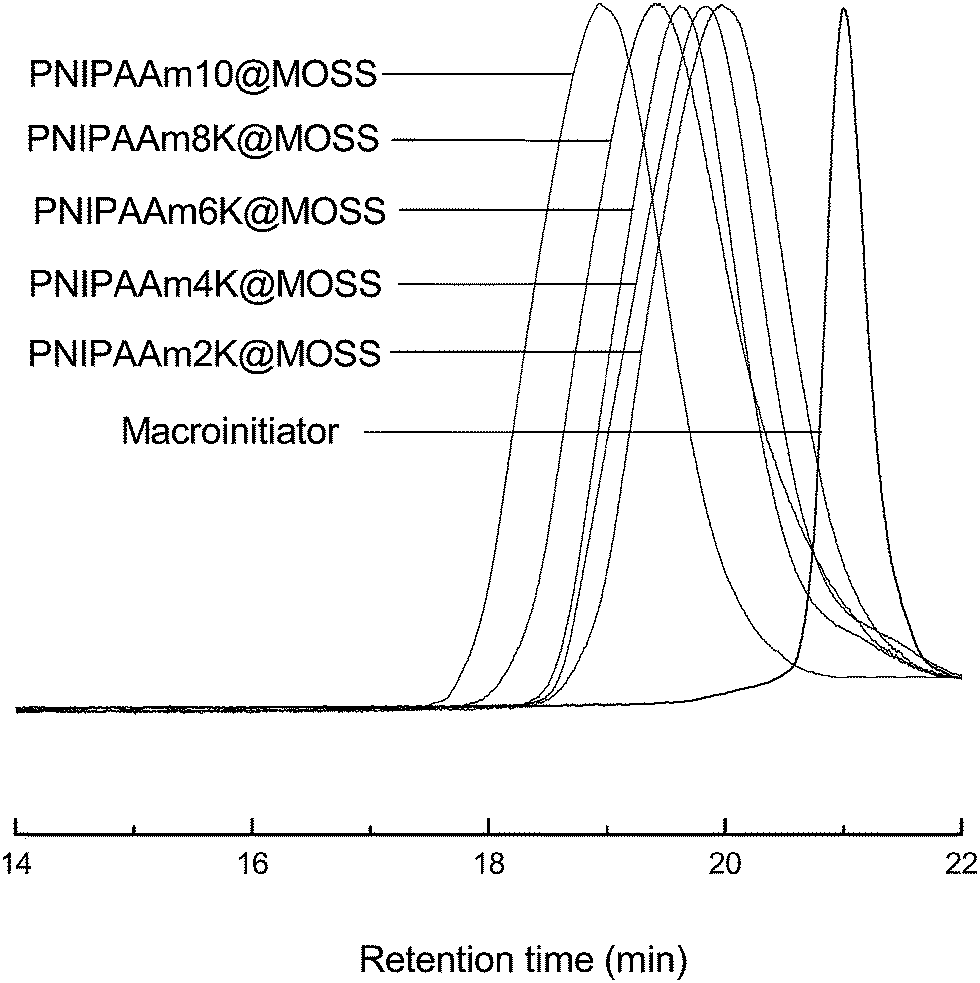
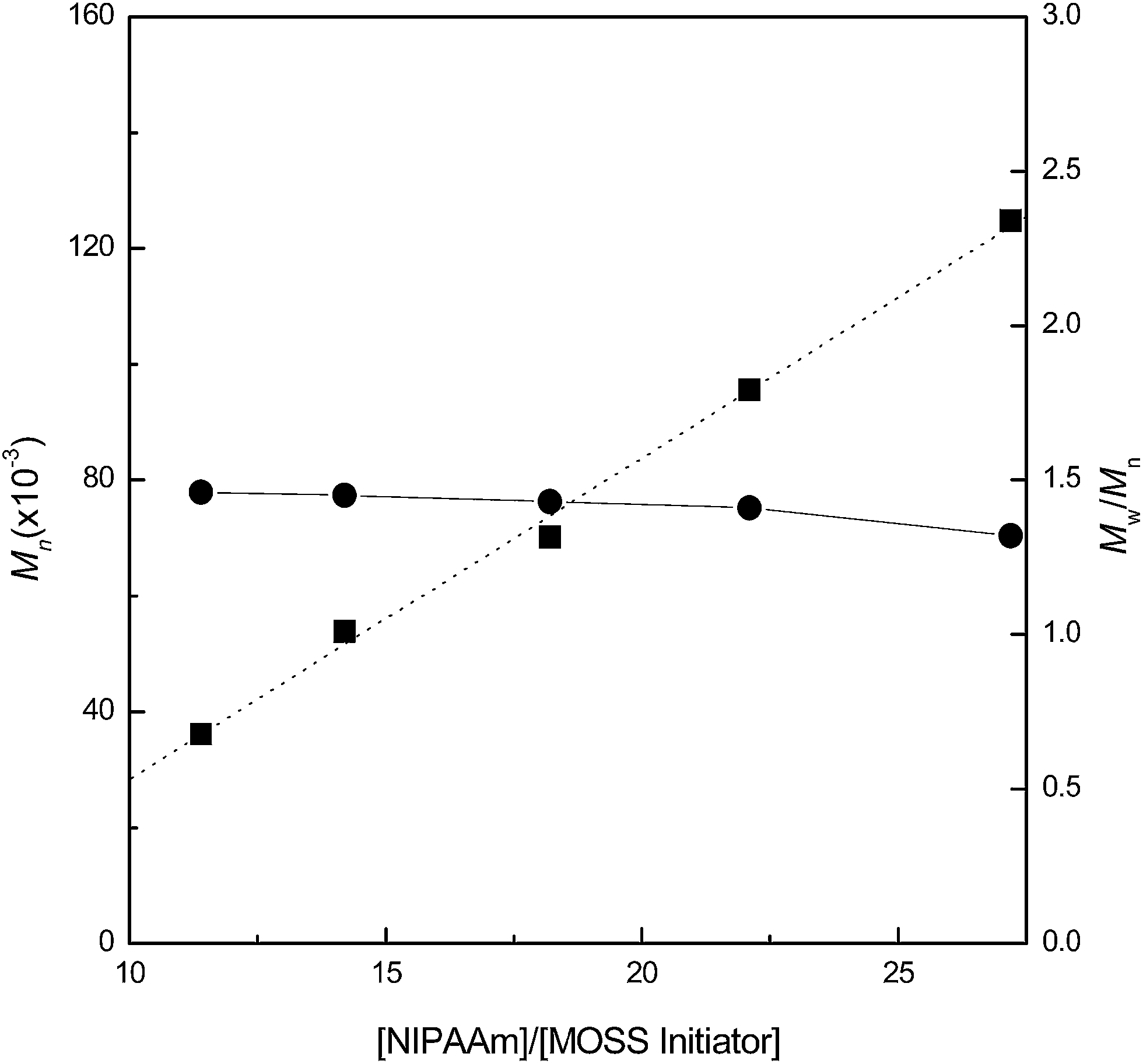
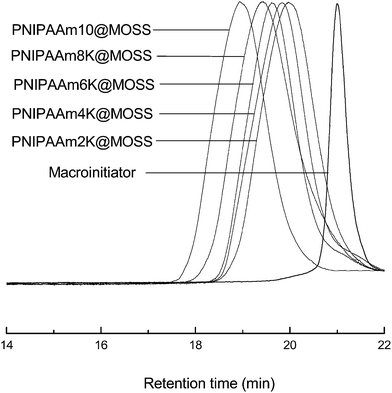
The MOSS macromer bearing 2-chloropropionate moieties was used as the macromolecular initiator, and the atom transfer radical polymerization (ATRP) was carried out to afford the organic–inorganic macromolecular brushes with MOSS backbone and with poly(N-isopropylacrylamide) (PNIPAAm) arms (see Scheme 1). By controlling the molar ratio of the macromolecular initiator to NIPAAm and the conversion of the monomer, the organic–inorganic molecular brushes with variable lengths of PNIPAAm were obtained. Representatively shown in Fig. 3 is the 1H NMR spectrum of PNIPAAm2K@MOSS. The signals of resonance at 1.14, 1.64, 2.02 and 4.01 ppm are assigned to the proton resonance of methyl, methylene, methine of PNIPAAm main chain, and methine of isopropyl groups of PNIPAAm, respectively; the broad signals of resonance at 6.0–7.0 ppm are assignable to the protons of N–H moiety. The signal of proton resonance at 0.10 ppm was discernable and is assignable to the protons of methyl groups of MOSS backbone. The 1H NMR spectra indicate that this product combined the structural features of PNIPAAm and MOSS. All PNIPAAm@MOSS samples with variable lengths of PNIPAAm chains together with compound 3 (i.e. macroinitiator) were subjected to gel permeation chromatography (GPC) to measure the molecular weights; the GPC profiles are presented in Fig. 4. The results of the molecular weights are summarized in Table 1. In all cases, unimodal peaks were displayed and no MOSS macromolecular initiator was detected in the resulting samples. Shown in Fig. 5 are the plots of number-average molecular weights and the values of polydispersity index as functions of the molar ratio of NIPAAm to the MOSS macromolecular initiator. The molecular weights increased almost linearly with increase of the molar ratio of NIPAAm to the MOSS macromolecular initiator; the polydispersity indices of PNIPAAm@MOSS were in the range of 1.30–1.46. The unimodal and fairly narrow distribution of molecular weights indicates that the MOSS macroinitiator (compound 3) can be used to obtain the PNIPAAm@MOSS samples, i.e., the organic–inorganic molecular brushes were successfully obtained, and that the polymerizations of NIPAAm with the MOSS macromolecular imitator proceeded in a living and controlled manner. It is noted that while the PNIPAAm arms were short (e.g., PNIPAAm2K@MOSS and PNIPAAm4K@MOSS), the distribution of the molecular weights was relatively broad. This observation could account for the restriction of the MOSS macrocycle on the ATRP of NIPAAm. In this work, the lengths of PNIPAAm arms were also estimated by thermogravimetric analysis (TGA). Assuming that the ceramic residues of the degradation were from MOSS, the lengths of PNIPAAm arms were thus estimated according to the yields of degradation (the TGA curves are not shown for brevity), and the results were also incorporated into Table 1. The values of molecular weights by GPC were lower than those estimated by TGA. This observation could be ascribed to the relatively low hydrodynamic volumes of the brush-like PNIPAAm samples.
 |
| | Fig. 4 GPC profiles of the organic–inorganic macrocyclic molecular brushes. | |
Table 1 Results of polymerization for the preparation of organic–inorganic molecular brushes composed of MOSS and PNIPAAm
| Sample |
Larm (PNIPAAm) |
Mna (Da) |
Mw/Mn |
Mnb (Da) |
| The values measured by means of gel permeation chromatography (GPC). The values estimated according to thermogravimetric analysis (TGA). |
| PNIPAAm10K@MOSS |
10100 |
124800 |
1.30 |
126200 |
| PNIPAAm8K@MOSS |
7700 |
95600 |
1.41 |
113560 |
| PNIPAAm6K@MOSS |
5600 |
70200 |
1.43 |
72800 |
| PNIPAAm4K@MOSS |
4300 |
54900 |
1.45 |
66720 |
| PNIPAAm2K@MOSS |
2700 |
36200 |
1.46 |
42460 |
| Macroinitiator |
— |
3400 |
1.12 |
— |
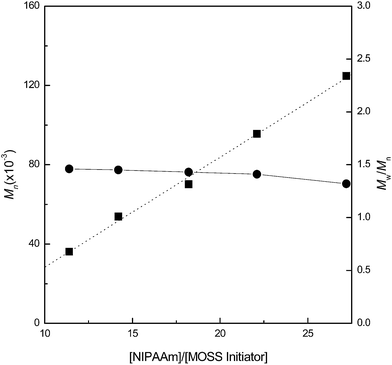 |
| | Fig. 5 Plots of number-average molecular weights and polydispersity indices as functions of the molar ratio of NIPAAm to the MOSS macromolecular initiator (3). | |
LCST behavior
PNIPAAm chains are capable of undergoing a coil-to-globule transition in aqueous solution in the vicinity of its lower critical solution temperature (LCST) at ∼32 °C.7–11 A single PNIPAAm chain adopts a random coil conformation in aqueous solution below the LCST. Upon heating up to the LCST or above, the random coil collapses into a globule owing to dehydration. In the present case, each PNIPAAm chain was grafted onto a bulky and hydrophobic MOSS backbone and possessed only one free end. It is of interest to examine the effect of the macrocyclic brush architecture on the LCST behavior of PNIPAAm chains. The thermoresponsive behavior was investigated by means of micro-differential scanning calorimetry (Micro-DSC) and UV-vis spectroscopy. Shown in Fig. 6 are the Micro-DSC curves of the aqueous solutions of the organic–inorganic molecular brushes. In the Micro-DSC curves, plain PNIPAAm displayed a single exothermic transition at 33.3 °C, attributable to the lower critical solution temperature (LCST) of this polymer. The exothermic transition stemmed from the coil-to-globule transition of the hydrated PNIPAAm chains. Notably, all PNIPAAm@MOSS samples displayed similar exothermic transitions. These exothermic peaks were asymmetrical with steep leading regions before approaching the minima. It is noted that the LCSTs of all organic–inorganic molecular brushes were significantly lower than that of plain PNIPAAm. The decreased LCSTs are attributable to the inclusion of the hydrophobic backbones (viz. MOSS), which reduced the affinity of PNIPAAm chains with water molecules. It is seen that the LCSTs of the hybrid molecular brushes decreased with decreasing length of the PNIPAAm chains. Also shown in Fig. 6 is the plot of the exothermic enthalpy (ΔHLCST) as a function of mass percentage of MOSS (see the inset). Assuming that the value of LCST enthalpy (denoted by ΔHLCST) is proportional to the quantity of the PNIPAAm chains which underwent the coil-to-globule transition, a straight line should be drawn according to the mass fraction of PNIPAAm chains if the LCST behavior of PNIPAAm chains were not affected by MOSS. However, the experimental ΔHLCST deviated negatively from this straight line. This observation suggests that the fractions of the PNIPAAm chains which can undergo the coil-to-globule transition in the organic–inorganic molecular brushes were significantly smaller than plain PNIPAAm. It is proposed that owing to the restriction of MOSS those PNIPAAm segments closely connected to MOSS macrocycles were unable to undergo the coil-to-globule transition: these PNIPAAm segments remained hydrophilic even while the solutions were heated to above the LCST. This speculation can be confirmed by means of cloud-point analysis and DLS (see infra).
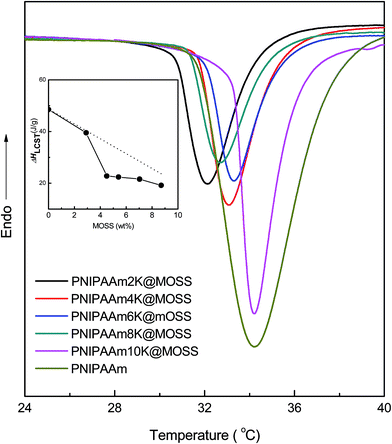 |
| | Fig. 6 Micro-DSC curves of the aqueous solutions of plain PNIPAAm (Mn = 12400 with Mw/Mn = 1.26) and the organic–inorganic macrocyclic molecular brushes. | |
The LCST behavior of the organic–inorganic PNIPAAm molecular brushes was further investigated in terms of cloud-point analysis by means of UV-vis spectroscopy. Shown in Fig. 7 are the plots of visible light transmittance for the organic–inorganic molecular brushes as function of temperature. For comparison, the curve of plain PNIPAAm (Mn = 12400 Mw/Mn = 1.26) aqueous solution was included. At low temperatures, the optical transmittance of all the aqueous solutions was as high as 97%. It is noted that the optical transmittance of the PNIPAAm@MOSS suspension was slightly lower than that of plain PNIPAAm. Upon heating the aqueous solutions to specific temperatures, the optical transmittance decreased significantly, indicating the occurrence of coil-to-globule transitions. For plain PNIPAAm, the LCST was measured to be ∼32 °C, and the transition range of temperature was as narrow as 2–3 °C. After the LCST the optical transmittance were decreased to ∼50% at 44 °C. Nonetheless, the PNIPAAm@MOSS suspensions displayed features quite different from those of plain PNIPAAm. First, the LCSTs of the PNIPAAm@MOSS samples were much lower than those of plain PNIPAAm; the LCSTs decreased with decreasing lengths of the PNIPAAm chains in the organic–organic molecular brushes. This observation was in good agreement with the Micro-DSC results. Second, the coil-to-globule transition ranges of the PNIPAAm@MOSS samples were significantly broadened, especially for the sample with shorter PNIPAAm chains (e.g., PNIPAAm2K@MOSS). The broadened coil-to-globule transition ranges could be associated with the restriction of MOSS on the coil-to-globule transitions of PNIPAAm chains. It is plausible to propose that the conformational alteration of PNIPAAm chains was hindered by the bulky MOSS backbone while the coil-to-globule transition of PNIPAAm side chains occurred. Third, the optical transmittance of all the organic–inorganic molecular brushes at elevated temperatures was significantly higher than that of the plain PNIPAAm. For instance, the optical transmittance of PNIPAAm10k@MOSS at 44 °C was still as high as 93% (see the inset, the optical images of the aqueous solution at 24 and 44 °C), suggesting that there was a considerable portion of the PNIPAAm chain which remained hydrated even at the temperature above the LCSTs. This observation can account for the restriction of MOSS on the coil-to-globule transition of PNIPAAm. This restriction resulted in a failure that the PNIPAAm segments became dehydrated above the LCST. The similar case has been theoretically rationalized by some investigators45,46 and has been found in organic PNIPAAm brushes.47–52
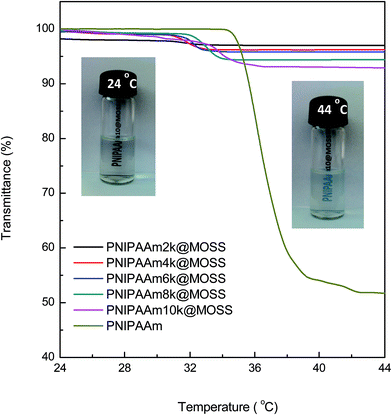 |
| | Fig. 7 Plots of light transmittance (λ = 550 nm) as functions of temperature for the aqueous solution (0.2 g L−1) of plain PNIPAAm and PNIPAAm@MOSS samples at a heating rate of 0.2 °C min−1. | |
Self-assembly behavior in bulk and aqueous solutions
The morphologies of the organic–inorganic molecular brushes in bulk were investigated by means of transmission electron microscopy (TEM). Shown in Fig. 8 are the TEM micrographs of PNIPAAm2K@MOSS, PNIPAAm4K@MOSS, PNIPAAm6K@MOSS, PNIPAAm8K@MOSS and PNIPAAm10K@MOSS. Notably, microphase-separated morphologies were exhibited in all cases. Spherical microdomains with the diameter of 10–50 nm were dispersed in a continuous matrix depending on the length of PNIPAAm chains. According to the difference in electron density between MOSS and PNIPAAm chains, the spherical microdomains are assignable to MOSS aggregates, whereas the continuous matrix is assignable to PNIPAAm. Notably, the size of MOSS microdomains decreased with increase in the lengths of PNIPAAm. The formation of the microphase-separated morphologies is attributable to the immiscibility of the inorganic backbone (viz. MOSS) with PNIPAAm chains; the spherical MOSS microdomains were formed via MOSS–MOSS interactions.
 |
| | Fig. 8 TEM micrographs of the organic–inorganic macrocyclic molecular brushes in bulk: (A) PNIPAAm10K@MOSS, (B) PNIPAAm8K@MOSS, (C) PNIPAAm6K@MOSS, (D) PNIPAAm4K@MOSS and (E) PNIPAAm2K@MOSS. | |
In view of the hydrophobicity of MOSS backbone and the water solubility of PNIPAAm chains, it is expected that the organic–inorganic molecular brushes would exhibit self-assembly behavior in aqueous solutions. The self-assembly behavior of the organic–inorganic molecular brushes was investigated by transmission electron microscopy (TEM) and dynamic light scattering (DLS). Shown in Fig. 9 are the TEM micrographs of the aqueous solutions after freeze drying. In the aqueous solutions, PNIPAAm2K@MOSS and PNIPAAm4K@MOSS were self-assembled into worm-like and interconnected nano-objects (Fig. 9A and B). With increasing lengths of the PNIPAAm chains, the organic–inorganic molecular brushes were gradually self-assembled into the spherical nano-objects 100–150 nm in size (see Fig. 9C–E). Shown in Fig. 10 are the plots of hydrodynamic radius distribution as function of hydrodynamic radius (Rh) for the aqueous solutions of the PNIPAAm@MOSS samples at 24 and 48 °C. Notably, all the organic–inorganic macrocyclic molecular brushes were capable of self-organizing into nano-objects in aqueous solution. Apart from PNIPAAm2K@MOSS, the intensity-averaged hydrodynamic radius (Rh) of all other PNIPAAm@MOSS samples displayed unimodal distribution dependent on the length of the PNIPAAm arms at 24 °C. Depending on the lengths of the PNIPAAm arms, the values of intensity-averaged hydrodynamic radius (Rh) were 239, 231, 226, 194 and 162 nm for PNIPAAm2K@MOSS, PNIPAAm4K@MOSS, PNIPAAm6K@MOSS, PNIPAAm8K@MOSS and PNIPAAm10K@MOSS at 24 °C, respectively; the Rh values decreased with increasing PNIPAAm arm length. It is proposed that the self-assembled nano-objects are micelle-like aggregates composed of hydrophobic cores via the MOSS–MOSS interactions and hydrated PNIPAAm coronas. It should be pointed out that for the aqueous solution containing PNIPAAm2K@MOSS, there were nano-objects with smaller size (i.e., Rh = 32 nm). The bimodal distribution of the hydrodynamic radius could result from the hydrophilicity/hydrophobicity balance not being fully established under the present conditions when the percentage of the inorganic hydrophobic component (viz. MOSS–MOSS) was too high. Furthermore, the results of DLS were in good agreement with those of TEM. It is should be pointed out that the sizes of aggregates measured with TEM (Fig. 9) were not necessarily identical to the Rh values by means of DLS. The former were obtained in the dry state and were much lower than the latter obtained in solution, which contains the dominant contribution of the solvated coronas (viz. PNIPAAm).
 |
| | Fig. 9 TEM micrographs of the organic–inorganic macrocyclic molecular brushes in aqueous solutions at 0.2 g L−1: (A) PNIPAAm2K@MOSS, (B) PNIPAAm4K@MOSS, (C) PNIPAAm6K@MOSS, (D) PNIPAAm8K@MOSS and (E) PNIPAAm10K@MOSS. | |
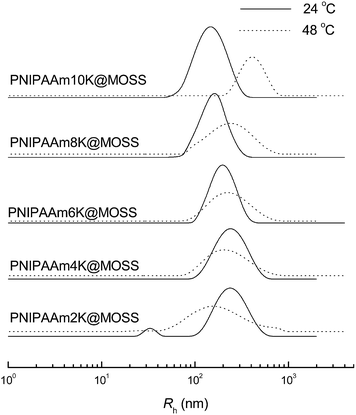 |
| | Fig. 10 Hydrodynamic radius (Rh) of PNIPAAm@MOSS samples in aqueous solutions (0.2 g L−1) at 24 (solid lines) and 48 °C (dashed lines). | |
Returning to Fig. 10, the coil-to-globule transition restriction of MOSS on PNIPAAm chains can be accounted for by the hydrodynamic radii at a temperature higher than that of the LCSTs (e.g., 48 °C). At 48 °C, the Rh values of PNIPAAm10K@MOSS, PNIPAAm8K@MOSS and PNIPAAm6K@MOSS were measured to be 412, 246 and 224 nm, respectively. The increased Rh values are readily interpretable on the basis of the occurrence of the coil-to-globule transition of PNIPAAm chains. With this transition, a portion of PNIPAAm chains became dehydrated. To establish a new hydrophobic/hydrophilic balance even at the elevated temperature, the organic–inorganic molecular brushes were self-organized into bigger nano-objects. For PNIPAAm6K@MOSS, PNIPAAm4K@MOSS and PNIPAAm2K@MOSS, however, the Rh values remained unchanged or even decreased, suggesting that in these systems, a considerable amount of PNIPAAm remains hydrated at this temperature. Owing to the presence of these hydrated PNIPAAm chains, the structural arrangement of the original aggregates resulted in the decreased size of the nano-objects under the new hydrophilic/hydrophobic balance. Notably, although decreased Rh values were observed for PNIPAAm4K@MOSS and PNIPAAm2K@MOSS at 48 °C, the distribution of Rh became fairly broad, possibly due to the restriction of MOSS on PNIPAAm chains.
Conclusions
Organic–inorganic molecular brushes composed of macrocyclic oligomeric silsesquioxanes (MOSS) and poly(N-isopropylacrylamide) (PNIPAAm) chains were synthesized via atom transfer radical polymerization (ATRP) with a 24-membered MOSS macromer bearing twelve 2-chloropropionate moieties. Gel permeation chromatography (GPC) showed that the polymerization of N-isopropylacrylamide (NIPAAm) mediated by copper(I) chloride and tris(2-(dimethylamino)ethyl)amine (Me6TREN) occurred in a living and controlled manner. Transmission electron microscopy (TEM) showed that the organic–inorganic molecular brushes were microphase-separated. The self-assembly of the organic–inorganic molecular brushes in bulk resulted in the formation of a morphology in which spherical MOSS microdomains with the diameter of 10–50 nm were dispersed into a continuous PNIPAAm matrix. All the organic–inorganic molecular brushes can be dispersed in water. The PNIPAAm@MOSS samples were capable of self-organizing into nano-objects in aqueous solutions, as evidenced by dynamic light scattering (DLS) and transmission election microscopy (TEM). The size of the nano-objects was quite dependent on the percentage of MOSS in the organic–inorganic molecular brushes. Both micro-differential scanning calorimetry (Micro-DSC) and ultraviolet-visible spectroscopy showed that the organic–inorganic molecular brushes can exhibit the thermoresponsive properties in aqueous solution as a plain PNIPAAm homopolymer. Nonetheless, the MOSS cages in the molecular brushes exerted a significant restriction on coil-to-globule transition of PNIPAAm chains.
Acknowledgements
The financial support from Natural Science Foundation of China (no. 21274091 and 51133003) was gratefully acknowledged.
References
- Y. C. Wang, L. Tang, Y. Li and J. Wang, Biomacromolecules, 2008, 10, 66 CrossRef PubMed.
- A. W. York, S. E. Kirkland and C. L. McCormick, Adv. Drug Delivery Rev., 2008, 60, 1018 CrossRef CAS PubMed.
- Y. Hu, Y. Chen, Q. Chen, L. Y. Zhang, X. Q. Jiang and C. Z. Yang, Polymer, 2005, 46, 12703 CrossRef CAS PubMed.
- Z. L. Ding, R. B. Fong, C. J. Long, P. S. Stayton and A. S. Hoffman, Nature, 2001, 411, 59 CrossRef CAS PubMed.
- Y. F. Zhang, S. Z. Luo and S. Y. Liu, Macromolecules, 2005, 38, 9813 CrossRef CAS.
- P. De, S. R. Gondi, D. Roy and B. S. Sumerlin, Macromolecules, 2009, 42, 5614 CrossRef CAS.
- M. Heskins and J. E. Guillet, J. Macromol. Sci., Part A, 1968, 2, 441 Search PubMed.
- P. Kujawa, F. Tanaka and F. M. Winnik, Macromolecules, 2006, 39, 3048 CrossRef CAS.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163 CrossRef CAS.
- C. Wu, Macromolecules, 1997, 30, 574 CrossRef CAS.
- P. Kujawa and F. M. Winnik, Macromolecules, 2001, 34, 4130 CrossRef CAS.
- Y. Hirokawa and T. Tanaka, J. Chem. Phys., 1984, 81, 6379 CrossRef PubMed.
- E. S. Matsuo and T. Tanaka, J. Chem. Phys., 1988, 89, 1695 CrossRef CAS PubMed.
- A. Suzuki and T. Tanaka, Nature, 1990, 346, 345 CrossRef CAS.
- K. Otake, H. Inomata, M. Konno and S. Saito, Macromolecules, 1990, 23, 283 CrossRef CAS.
- H. Inomata, S. Goto, K. Otake and S. Saito, Langmuir, 1992, 8, 687 CrossRef CAS.
- M. Shibayama, M. Morimoto and S. Nomura, Macromolecules, 1994, 27, 5060 CrossRef CAS.
- L. Liang, P. C. Rieke, J. Liu, G. E. Fryxell, J. S. Young, M. H. Engelhard and K. L. Alford, Langmuir, 2000, 16, 8016 CrossRef CAS.
- J. Shan, J. Chen, M. Nuopponen and H. Tenhu, Langmuir, 2004, 20, 4671 CrossRef CAS.
- M. Nuopponen, J. Ojala and H. Tenhu, Polymer, 2004, 45, 3643 CrossRef CAS PubMed.
- X. Zhou, X. Ye and G. Zhang, J. Phys. Chem. B, 2007, 111, 5111 CrossRef CAS PubMed.
- J. Mu and S. Zheng, J. Colloid Interface Sci., 2007, 307, 377 CrossRef CAS PubMed.
- K. Zeng, Y. Fang and S. Zheng, J. Polym. Sci., Part B: Polym. Phys., 2009, 47, 504 CrossRef CAS.
- W. Zhang, L. Liu, X. Zhuang, X. Li, J. Bai and Y. Chen, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7049 CrossRef CAS.
- Y. Zheng, L. Wang and S. Zheng, Eur. Polym. J., 2012, 48, 945 CrossRef CAS PubMed.
- L. Wang, K. Zeng and S. Zheng, ACS Appl. Mater. Interfaces, 2011, 3, 898 CAS.
- S. W. Kuo, J. L. Hong, Y. C. Huang, J. K. Chen, S. K. Fan, F. H. Ko, C. W. Chu and F. C. Chang, J. Nanomater., 2012, 749732, DOI:10.1155/2012/749732.
- Y. Bai, J. Wei, L. Yang, C. He and X. Lu, Colloid Polym. Sci., 2012, 290, 507 CAS.
- G. Li, L. Wang, H. Ni and C. U. Pittman, J. Inorg. Organomet. Polym., 2001, 11, 123 CrossRef CAS.
- O. I. Shchegolikhina, V. A. Igonin, Y. A. Molodtsova and Y. A. Pozdnyakova, J. Organomet. Chem., 1998, 562, 141 CrossRef CAS.
- A. A. Zhdanov and T. V. Strelkova, J. Organomet. Chem., 1998, 562, 141 CrossRef.
- N. V. Sergienko, E. S. Trankina, V. I. Pavlov, A. A. Zhdanov, K. A. Lyssenko and M. Y. Antipin, Russ. Chem. Bull., Int. Ed., 2004, 53, 351 CrossRef CAS.
- Y. A. Molodtsova, Y. A. Pozdnyakova, K. A. Lyssenko, I. V. Blagodatskikh, D. E. Katsoulis and O. I. Shchegolikhina, J. Organomet. Chem., 1998, 571, 31 CrossRef CAS.
- O. I. Shchegolikhina, Y. A. Pozdnyakova, S. D. Molodtsova and B. Korkin, Organometallics, 2000, 19, 1077 CrossRef CAS.
- O. I. Shchegolikhina, Y. A. Pozdnyakova, Y. A. Molodtsova, S. D. Korkin, S. S. Bukalov, L. A. Leites, K. A. Lyssenko, A. S. Peregudov, N. Auner and D. E. Katsoulis, Inorg. Chem., 2002, 41, 6892 CrossRef CAS PubMed.
- Y. A. Pozdnyakova, K. A. Lyssenko, I. V. Blagodatskikh, N. Auner, D. E. Katsoulis and O. I. Shchegolikhina, Eur. J. Inorg. Chem., 2004, 2004, 1253 CrossRef.
- O. I. Shchegolikhina, Y. A. Pozdnyakova, M. Y. Antipin, D. E. Katsoulis, N. Auner and B. Herrschaft, Organometallics, 2000, 19, 1077 CrossRef CAS.
- J. Han and S. Zheng, Macromolecules, 2008, 41, 4561 CrossRef CAS.
- J. Han and S. Zheng, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6894 CrossRef CAS.
- J. Han, L. Zhu and S. Zheng, Eur. Polym. J., 2012, 48, 730 CrossRef CAS PubMed.
- L. Zhu, C. Zhang, J. Han and S. Zheng, Soft Matter, 2012, 8, 7062 RSC.
- J. S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614 CrossRef CAS.
- Y. A. Molodtsova, K. A. Lyssenko, I. V. Blagodatskikh, E. V. Matukhina, A. S. Peregudov, M. I. Buzin, V. G. Vasil’ev, D. E. Katsoulis and O. I. Shchegolikhina, J. Organomet. Chem., 2008, 693, 1797 CrossRef CAS PubMed.
- Y. A. Molodtsova, Y. A. Pozdnyakova, I. V. Blagodatskikh, A. S. Peregudov and O. I. Shchegolikhina, Russ. Chem. Bull., Int. Ed., 2003, 52, 2722 CrossRef CAS.
- E. B. Zhulina, O. V. Borisov, V. A. Pryamitsyn and T. M. Birshtein, Macromolecules, 1991, 24, 140 CrossRef CAS.
- G. S. Grest and M. Murat, in Monte Carlo and Molecular Dynamics Simulations in Polymer Science, ed. K. Binder, Clarendon, Oxford, 1994 Search PubMed.
- G. E. Yakubov, B. Loppinet, H. Zhang, J. Rühe, R. Sigel and G. Fytas, Phys. Rev. Lett., 2004, 92, 115501 CrossRef CAS.
- A. Domack, S. Prucker, J. Ruhe and D. Johannsmann, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1997, 56, 680 CrossRef CAS.
- A. Karim, S. K. Satija, J. F. Douglas, J. F. Ankner and L. J. Fetters, Phys. Rev. Lett., 1994, 73, 3407 CrossRef CAS.
- S. Balamurugan, S. Mendez, S. S. Balamurugan, M. J. I. I. O’ Brien and G. P. Lopez, Langmuir, 2003, 19, 2545 CrossRef CAS.
- H. Yim, M. S. Kent, S. Mendez, S. S. Balamurugan, S. Balamurugan, G. P. Lopez and S. Satija, Macromolecules, 2004, 37, 1994 CrossRef CAS.
- P. W. Zhu and D. H. Napper, Langmuir, 1996, 12, 5992 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.