
Mechanically strong and stretchable polyurethane–urea supramolecular hydrogel using water as an additional in situ chain extender†
Abstract
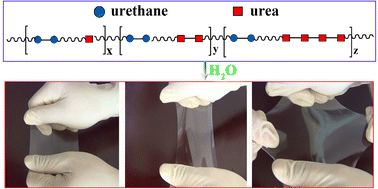
Mechanically strong hydrogels have attracted much interest as a result of their potential applications as biomaterials. However, it is still a challenge to produce mechanically strong supramolecular hydrogels because of the inherently weak characteristics of non-covalent interactions. A novel polyurethane–urea supramolecular hydrogel with excellent mechanical properties was developed in our laboratory by chance during the preparation of a water-borne dispersion of polyurethane with an excess amount of –NCO groups. Subsequent studies showed that this mechanical strength was because of the slow formation of multi-urea linkages and further chain extension because of the reaction of water with the excess –NCO groups in the isocyanate prepolymer chains or the free diisocyanate, or both. The mechanical properties of the polyurethane–urea supramolecular hydrogels obtained can be adjusted by simply altering the diisocyanate content. The following ranges of properties were obtained: shear modulus, 0.2–0.8 MPa; elongation at breakage, 970–2420%; tensile strength, 3.3–34 MPa; and compression stress, up to 38 MPa. Further analysis showed that the elongation ratio and tensile stress at breakage linearly decreased and increased, respectively, with an increase in the ratio of the hard segment.
 Please wait while we load your content...
Please wait while we load your content...

