
Phosphate-containing metabolites switch on phosphorescence of ferric ion engineered carbon dots in aqueous solution†
Abstract
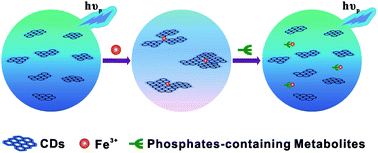
While most research has focused on the development of carbon dot (CD) based fluorescence sensors, much less attention has been paid to the phosphorescence phenomenon and its potential applications to date. Herein, room temperature phosphorescence (RTP) of water soluble CDs free of deoxidants and other inducers was observed for the first time in pure aqueous solution. RTP of CDs could be significantly quenched when chelating with iron ions as well as aggregation of CDs, presumably resulting from the formation of non-luminescent chelate. Due to a high affinity of iron ions to phosphate ions through well-known Fe–O–P bonds, the quenched RTP of functionalized CDs by Fe3+ could be basically recovered in the presence of phosphate-containing molecules. For a proof-of-concept demonstration, adenosine-5′-triphosphate (ATP), as a common phosphate-containing metabolite was quantitatively detected by a phosphorescence “off-to-on” approach. The enhancement of RTP at 440 nm was linearly proportional to the concentrations of ATP ranging from 20 to 200 μM with a detection limit as low as 14 μM. Moreover, the iron ion engineered CDs based RTP probe was used to estimate ATP levels in human blood plasma.
 Please wait while we load your content...
Please wait while we load your content...

