Copper-catalyzed N-arylation of azoles and diazoles using highly functionalized trivalent organobismuth reagents†
Pauline Petiot,
Julien Dansereau and
Alexandre Gagnon*
Département de chimie, Université du Québec à Montréal, C.P. 8888, Succ. Centre-Ville, Montréal, Québec H3C 3P8, Canada. E-mail: gagnon.alexandre@uqam.ca
First published on 18th April 2014
Abstract
The N-arylation of indoles, indazoles, pyrroles, and pyrazoles using highly functionalized trivalent arylbismuth reagents is reported. The reaction is promoted by a substoichiometric amount of copper acetate, and it tolerates a wide diversity of functional groups on the azole and the organobismuth reagent. The method is also applied to the N-arylation of tryptophan derivatives.
Azoles and diazoles are common scaffolds that are frequently used in medicinal chemistry to project key pharmacophores along different vectors inside the binding pocket of a biological target.1,2 N-Arylation of these nitrogenated compounds makes it possible to screen for new interactions around an inhibitor3 and to modify its biophysical properties.4 N-Arylazoles have also found numerous applications in material and polymer sciences.5 N-Arylazoles and diazoles are commonly prepared via metal-catalyzed N-arylation6 of N–H heteroarenes using aryl halides,7 arylboronic acids,8 or aryllead9 reagents. However, lead reagents are highly toxic, and other methods sometimes require extended reaction times, super-stoichiometric amounts of catalyst or costly ligands to obtain good yields. Consequently, new procedures that lead to the facile installation of densely functionalized aryl groups on azoles and diazoles are still desirable.
Over the past years, we have disclosed a portfolio of reactions for the formation of C–C,10 C–N,11 and C–O12 bonds using organobismuth reagents. Organobismuthanes can be prepared from inexpensive and non-toxic bismuth salts and offer mild reactivity that tolerates the presence of numerous functional groups on both the substrate and the organometallic species.13 Barton and Finet reported in 1988 the use of triphenylbismuth-bis-trifluoroacetate in the arylation of indole A (Scheme 1).14 However, the method was applied exclusively to the transfer of an unsubstituted phenyl group and required the use of a pentavalent bismuth species, which is less stable than its trivalent counterpart. More importantly, C3-arylation was observed in most cases, except when the C3 position was blocked by an alkyl group or when an ester was present at the C2 position of indole.
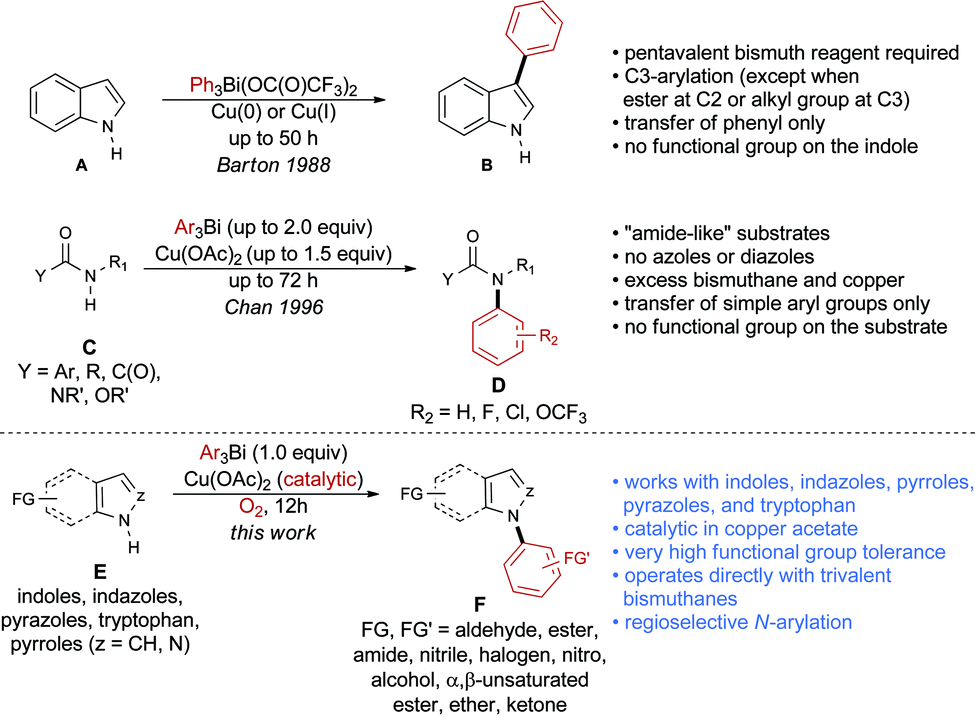 | ||
| Scheme 1 Comparison of our N-arylation reaction with precedents in the literature. | ||
In 1996, Chan published a variation of a protocol disclosed by Barton and Finet,15 in which trivalent bismuthanes are used to N-arylate nitrogenated compounds (Scheme 1).16 While this method is very useful, it suffers from multiple limitations as it requires up to 2.0 equivalents of the organobismuth reagent, 1.5 equivalents of the copper catalyst and reaction times that are as long as 72 hours. Moreover, the method was not applied to indoles, indazoles, pyrazoles, or pyrroles, but rather to type C compounds, in which the nitrogen atom is connected to a carbonyl functional group. Finally, functional group tolerance was not demonstrated, as only simple arylbismuthanes were coupled with substrates bearing no functionalities.
We report herein the first method for the N-arylation of azoles and diazoles E using highly functionalized trivalent bismuth reagents and promoted by catalytic amounts of copper acetate. The procedure exclusively generates the N-arylation product and shows exceptional functional group tolerance on both coupling partners. The procedure is also applied to the N-arylation of tryptophan derivatives to afford N-arylindolyl products.
We began by optimizing the conditions for the phenylation of methyl indole-5-carboxylate 1 using the Barton–Finet–Chan protocol (Table 1).15,16 Upon reacting 1.0 equivalent of triphenylbismuth with indole 1 in the presence of 1.5 equivalents of copper acetate and 3.0 equivalents of pyridine under air, we obtained the N-arylated product 2 regioselectively in quantitative yield (entry 1). This result is interesting and important for two reasons. First, to the best of our knowledge, this is the first time that trivalent organobismuth reagents are used directly in the N-arylation of indoles, eliminating the need to prepare the less stable bis-trifluoroacetate species. Second, in contrast with triphenylbismuth-bis-trifluoroacetate, the process involving triphenylbismuth leads exclusively to N-arylation, as opposed to C3-arylation. Using 0.7 equivalent of the organobismuth species resulted in a 25% decrease in the yield of the reaction, suggesting that only one phenyl group can be transferred from the organobismuth species during the reaction (entry 2).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ph3Bi (x equiv.) | Cu(OAc)2 (y equiv.) | Base | Solvent | Atm. | Yielda (%) |
| a Isolated yield of pure product 2.b Reaction performed with 1.0 equiv. of base. | ||||||
| 1 | 1.0 | 1.5 | Pyridine | CH2Cl2 | Air | 99 |
| 2 | 0.7 | 1.5 | Pyridine | CH2Cl2 | Air | 74 |
| 3 | 1.0 | 1.0 | Pyridine | CH2Cl2 | Air | 45 |
| 4 | 1.0 | 1.0 | Et3N | CH2Cl2 | Air | 45 |
| 5 | 1.0 | 1.0 | Pyridine | CH3CN | Air | 76 |
| 6 | 1.0 | 1.0 | Pyridine | CH2Cl2 | O2 | 96 |
| 7 | 1.0 | 1.0 | Pyridine | CH2Cl2 | Ar | 47 |
| 8 | 1.0 | 0.1 | Pyridine | CH2Cl2 | O2 | 99 |
| 9 | 1.0 | 0.05 | Pyridine | CH2Cl2 | O2 | 45 |
| 10b | 1.0 | 0.1 | Pyridine | CH2Cl2 | O2 | 96 |
| 11 | 1.0 | 0.1 | Pyridine | THF | O2 | 82 |
| 12 | 1.0 | 0.1 | Pyridine | DME | O2 | 57 |
| 13 | 1.0 | 0.1 | Pyridine | CH3CN | O2 | 54 |
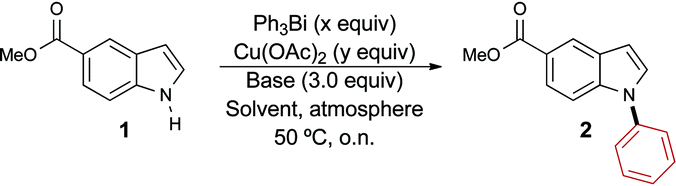
In order to develop a more efficient protocol, we next sought to reduce the catalyst loading. Unfortunately, the use of a stoichiometric amount of copper acetate led to a significant decrease in the yield of the reaction (entry 3), motivating us to revisit other parameters of the reaction. Changing the base to triethylamine proved inconsequential (entry 4), but an improvement in the yield of the reaction was observed on conducting the reaction in acetonitrile (entry 5). We recently reported the beneficial effect of oxygen during our studies on the O-arylation of phenols using trivalent organobismuthanes.12 Therefore, we next investigated the use of oxygen as the reaction atmosphere and observed a near quantitative yield for the formation of 2 (entry 6). To further validate the importance of oxygen, we performed the reaction under argon and observed a yield similar to when the reaction was run under ambient air, thus confirming the positive effect of oxygen on the reaction (entry 7 vs. 3). Encouraged by this observation, we then gradually lowered the amount of catalyst and found that the yield was retained upon using 10 mol% of copper acetate (entry 8). However, reducing the loading further was not tolerated and produced only a modest yield of the desired N-phenyl indole 2 (entry 9). Although Barton reported that the exclusion of oxygen led to a negative impact on the arylation of amines with trivalent organobismuthanes,15 the possibility of lowering the copper acetate loading by performing the reaction under pure oxygen was not demonstrated. It is likely that the role of the oxygen is to oxidize the copper species with lower valency, which are generated during the reaction, to the +2 oxidation state. To minimize the amount of base, we performed the reaction using 1.0 equivalent of pyridine and still obtained a near quantitative yield of product 2 (entry 10). The exploration of non-halogenated solvents (entries 11–13) led to the identification of THF as the best alternative to dichloromethane. Using our optimal conditions, we next explored the impact of varying the steric and electronic properties of the organobismuthane on the yield of the arylation reaction. The functionalized organobismuthanes needed for this study (Fig. 1) were prepared according to procedures that we previously reported.10b,12
 | ||
| Fig. 1 Functionalized organobismuthanes 3a–q used in the N-arylation of azoles and diazoles. | ||
Our studies show that the transfer of para and meta-tolyl groups proceeds efficiently, delivering the corresponding products 4a and 4b in excellent yields (Scheme 2). The transfer of the more sterically hindered ortho-tolyl group proved more challenging and required a higher catalyst loading to provide the desired product 4c in an acceptable yield. While it is difficult to establish a clear correlation between the electronic properties of the organobismuthane and the yield of the arylation reaction, the results obtained to date demonstrate that good yields are afforded by using triarylbismuthanes substituted with electron-donating (4a, b, d) and electron-withdrawing (4e–h) groups. Interestingly, a 2,6-dimethylphenyl unit was installed on indole 1 using our conditions, although with a modest yield (compound 4i). Note that very few methods exist for the transfer of this highly hindered group.17
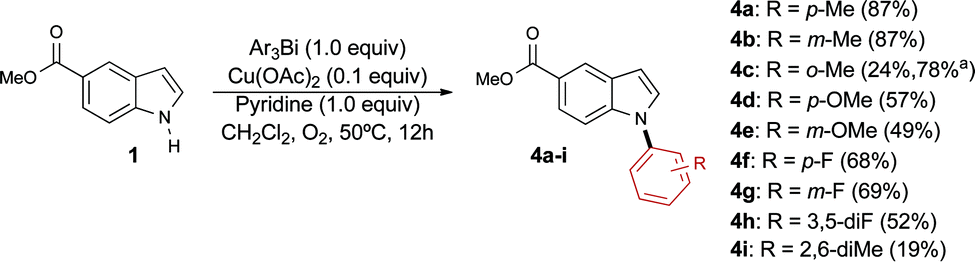 | ||
| Scheme 2 Steric and electronic effects of organobismuthane on the N-arylation of 1. a Alternative conditions were employed for compound 4c: Ar3Bi (1.0 equiv.), Cu(OAc)2 (1.0 equiv.), pyridine (3.0 equiv.), CH2Cl2, O2, 50 °C, 12 h. | ||
In order to further expand the functional group tolerance, we prepared three new bismuth reagents by performing a functional group manipulation directly on selected organobismuthanes. As illustrated in Scheme 3, triarylbismuthanes 3r and 3s, bearing alcohol functional groups, were synthesized by Grignard addition on the ester 3p and on the aldehyde 3m, respectively. These groups are important in medicinal chemistry as the presence of an alcohol group controls the lipophilicity (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D) of arylated products that are generated in the next step. A Horner–Emmons–Wadsworth reaction was also performed on 3m to furnish the cinnamyl ester 3t. This functional group is also frequently found in numerous bioactive compounds.
D) of arylated products that are generated in the next step. A Horner–Emmons–Wadsworth reaction was also performed on 3m to furnish the cinnamyl ester 3t. This functional group is also frequently found in numerous bioactive compounds.
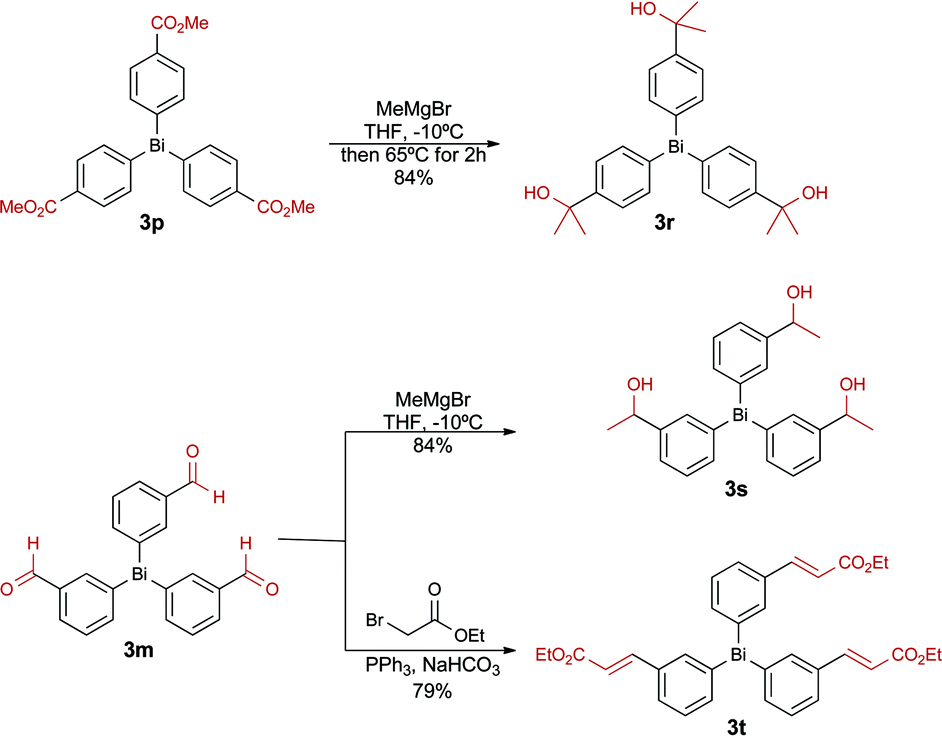 | ||
| Scheme 3 Preparation of highly functionalized organobismuthanes by functional group manipulation. | ||
The scope of the reaction was then studied by arylating different azoles and diazoles 5 using highly functionalized triarylbismuthanes 3a–t (Scheme 4). Our investigations reveal that the reaction proceeds smoothly on indoles (6a–o), pyrroles (6p, q), and pyrazoles18 (6r, s) to afford the desired N-arylated products in good to excellent yields. These results also suggest that the substitution pattern of the indole has little impact on the outcome of the reaction. However, using our conditions, we were not able to arylate 7-methylindole, possibly due to the development of a strong 1,3-allylic-type strain during the formation of the product. In the case of pyrazole 6s, the product of arylation on the nitrogen distal to the tolyl group was obtained, as confirmed by NMR studies. Importantly, the procedure tolerates a wide variety of functional groups on the azole such as aldehydes (6a–c), nitriles (6d–f), O-acetates (6g), nitro groups (6m), amides (6n, o), and ketones (6p, q). Note that bromides (6h, i), and iodides (6k), which could be expected to interfere in the direct N-arylation of azoles with aryl halides, were found to be inert under these conditions. Numerous functional groups can also be present on the organobismuth reagents, such as acetals (6a, m, n), fluorides (6b, c), aldehydes (6f, p), esters (6i), ethers (6q, s) and α,β-unsaturated esters (6k). Interestingly, we found that an alcohol can be present on the substrate (6j, l) or on the arylbismuth reagent (6r) without undergoing O-arylation; moreover, a secondary amide is not arylated under these conditions (6n). Lastly, a cyclopropylphenyl (6e) and a trifluoromethylphenyl (6g) group were efficiently transferred using this method. These groups are important in medicinal chemistry as they show increased metabolic stability compared to typical alkyl groups.19
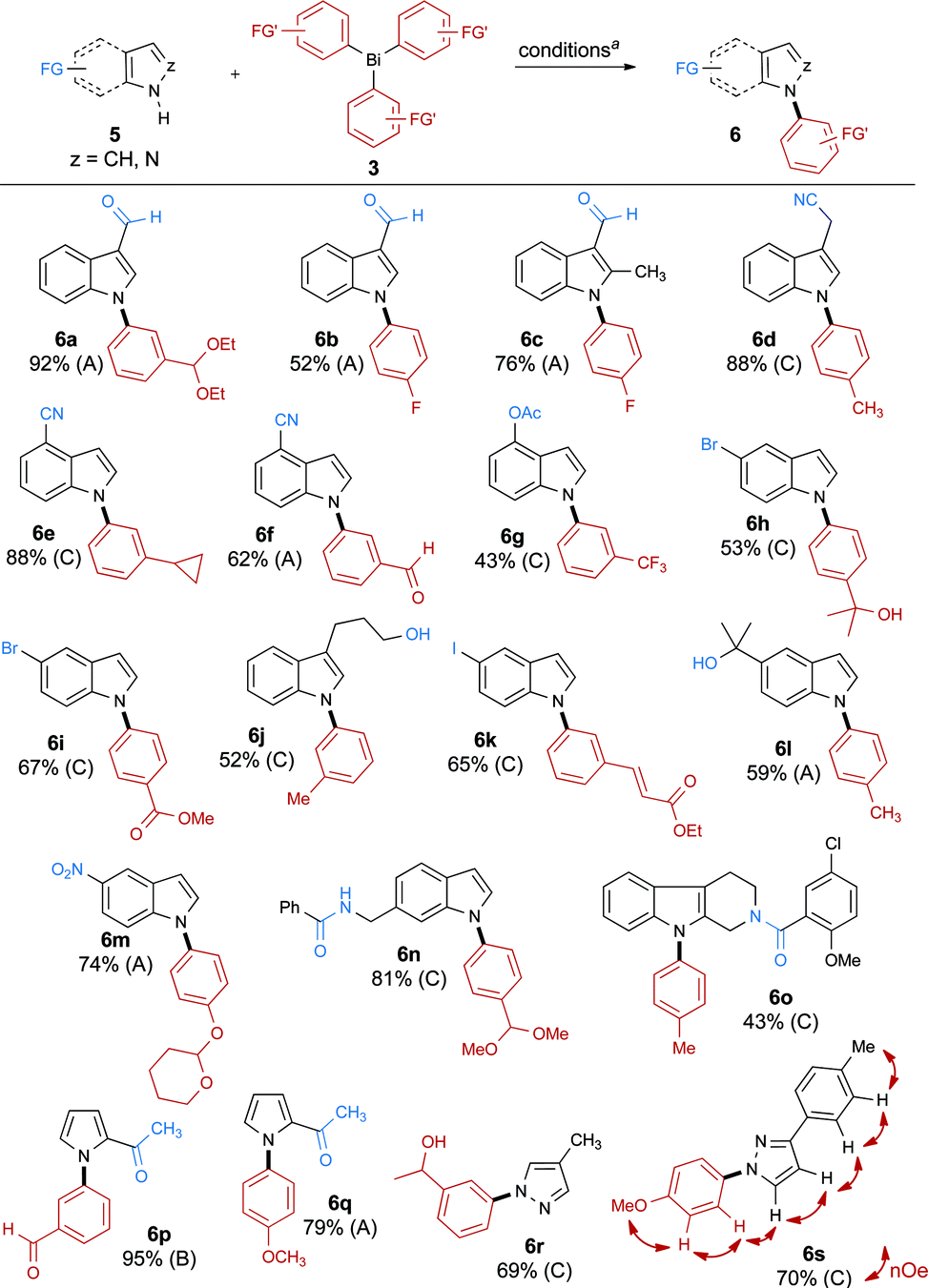 | ||
| Scheme 4 N-Arylation of azoles and diazoles 5 using functionalized triarylbismuthanes 3a–t. a Condition A: Ar3Bi (1.0 equiv.), Cu(OAc)2 (0.1 equiv.), pyridine (1.0 equiv.), CH2Cl2, O2, 50 °C; condition B: Ar3Bi (1.0 equiv.), Cu(OAc)2 (0.1 equiv.), pyridine (3.0 equiv.), CH2Cl2, O2, 50 °C; condition C: Ar3Bi (1.0 equiv.), Cu(OAc)2 (1.0 equiv.), pyridine (3.0 equiv.), CH2Cl2, O2, 50 °C. | ||
The N-arylation of indazoles was next evaluated, keeping in mind that a mixture of regioisomers resulting from arylation at N1 and N2 is often obtained with other methods.18,20 When 1H-indazole-6-carbaldehyde 7 was subjected to our conditions, the product 8a generated from the arylation of N1 was predominantly formed (Scheme 5). The structure of the minor regioisomer 8b obtained from the arylation of N2 was established by X-ray crystallography.
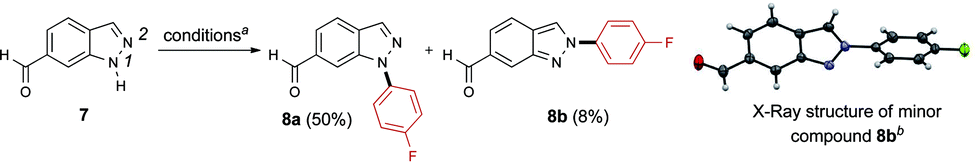 | ||
| Scheme 5 N-Arylation of 1H-indazole-6-carbaldehyde 7. a Conditions: (p-FC6H4)3Bi (1.0 equiv.), Cu(OAc)2 (1.0 equiv.), pyridine (3.0 equiv.), DCM, 50 °C, O2, o.n.; b ORTEP diagram of compound 8b: thermal ellipsoids are shown at the 50% probability level. | ||
Derivatives of tryptophan are important in medicinal chemistry as these species are involved in numerous biological processes and diseases.21 The post-synthetic modification of peptides containing tryptophan residues has also found applications in chemical biology.22 To the best of our knowledge, very few methods for the N-arylation of tryptophan derivatives have been reported in the literature.23 Notably, Boc- and Fmoc-protected tryptophan derivatives 9a and 9b were smoothly N-arylated using our protocol to selectively provide the N-indolyl derivatives 10a, b in good yield (Scheme 6).24
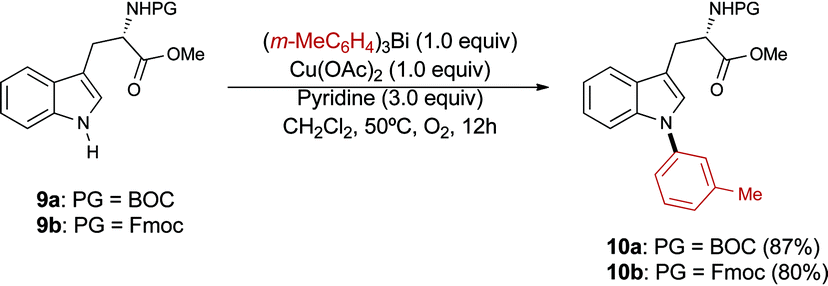 | ||
| Scheme 6 N-Arylation of tryptophan derivatives 9a, b. | ||
Conclusions
In summary, we have developed an efficient and general method for the N-arylation of indoles, indazoles, pyrroles, and pyrazoles that proceeds directly using highly functionalized trivalent organobismuth reagents. The transformation is promoted by catalytic amounts of copper acetate and tolerates an exceptional diversity of functional groups on both coupling partners, giving access to highly functionalized azoles. The protocol was also applied to the N-arylation of tryptophan derivatives. Application of this methodology to other arylation reactions, including amino-acids and tryptophan-containing peptides, is in progress in our laboratory and the results will be reported in due course.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Université du Québec à Montréal (UQÀM). We thank Pr. Xavier Ottenwaelder, Mohammad S. Askari (Concordia University), and Francine Bélanger-Gariépy (Université de Montréal) for the crystallographic analysis of compound 8b and Dr Alexandre Arnold (UQÀM) for NMR analysis of compound 6s. P.P. thanks UQÀM for a FARE scholarship. J.D. thanks the Faculty of Sciences of UQÀM for an undergraduate research scholarship. A.G. thanks Dr Hugo Lachance for useful discussions.Notes and references
- For reviews on pyrroles in medicinal chemistry, see: (a) U. Bhatt, B. C. Duffy, P. R. Guzzo, L. Cheng and T. Elebring, Synth. Commun., 2007, 37, 2793 CrossRef CAS; (b) V. Estévez, M. Villacampa and J. C. Menéndez, Chem. Soc. Rev., 2010, 39, 4402 RSC.
- For reviews on indoles and other azoles in medicinal chemistry, see: (a) V. Sharma, P. Kumar and D. Pathak, J. Heterocycl. Chem., 2010, 47, 491 CAS; (b) F. Rodrigues de Sa Alves, E. J. Barreiro and C. A. M. Fraga, Mini-Rev. Med. Chem., 2009, 9, 782 CrossRef CAS; (c) S. Biswal, U. Sahoo, S. Sethy, H. K. S. Kumar and M. Banerjee, Asian J. Pharm. Clin. Res., 2012, 5, 1 CAS; (d) N. K. Kaushik, N. Kaushik, P. Attri, N. Kumar, C. H. Kim, A. K. Verma and E. H. Choi, Molecules, 2013, 18, 6620 CrossRef CAS PubMed; (e) V. Kumar, K. Kaur, G. K. Gupta and A. K. Sharma, Eur. J. Med. Chem., 2013, 69, 735 CrossRef CAS PubMed; (f) X.-M. Peng, G.-X. Cai and C.-H. Zhou, Curr. Top. Med. Chem., 2013, 13, 1963 CrossRef CAS.
- C. Bissantz, B. Kuhn and M. Stahl, J. Med. Chem., 2010, 53, 5061 CrossRef CAS PubMed.
- G. M. Keserü and G. M. Makara, Nat. Rev. Drug Discovery, 2009, 8, 203 CrossRef PubMed.
- Selected examples of applications of N-arylazoles in material and polymer sciences: (a) C. A. Zuniga, J. Abdallah, W. Haske, Y. Zhang, I. Coropceanu, S. Barlow, B. Kippelen and S. R. Marder, Adv. Mater., 2013, 25, 1739 CrossRef CAS PubMed; (b) L. Xu, B. Zhang, Y. Chen, K.-G. Neoh, E.-T. Kang and G. D. Fu, Macromol. Rapid Commun., 2013, 34, 234 CrossRef CAS PubMed; (c) X. Liu, W. Wen and G. C. Bazan, Adv. Mater., 2012, 24, 4505 CrossRef CAS PubMed; (d) J. You, G. Li and Z. Wang, Polymer, 2012, 53, 5116 CrossRef CAS PubMed; (e) C.-N. Chuang, H.-J. Chuang, Y.-X. Wang, S.-H. Chen, J.-J. Huang, M.-k. Leung and K.-H. Hsieh, Polymer, 2012, 53, 4983 CrossRef CAS PubMed; (f) S. O. Jeon and J. Y. Lee, J. Mater. Chem., 2012, 22, 10537 RSC; (g) J. F. Lee, S. L. C. Hsu, P. I. Lee, H. Y. Chuang, M. L. Yang, J. S. Chen and W. Y. Chou, Sol. Energy Mater. Sol. Cells, 2011, 95, 2795 CrossRef CAS PubMed; (h) J. Li, Q. Li and D. Liu, ACS Appl. Mater. Interfaces, 2011, 3, 2099 CrossRef CAS PubMed; (i) Y.-H. Chen, Y.-Y. Lin, Y.-C. Chen, J. T. Lin, R.-H. Lee, W.-J. Kuo and R.-J. Jeng, Polymer, 2011, 52, 976 CrossRef CAS PubMed; (j) H.-Y. Chen, C.-T. Chen and C.-T. Chen, Macromolecules, 2010, 43, 3613 CrossRef CAS; (k) K. Zhang, Y. Tao, C. Yang, H. You, Y. Zou, J. Qin and D. Ma, Chem. Mater., 2008, 20, 7324 CrossRef CAS; (l) Z. Huang, J.-H. Ryu, E. Lee and M. Lee, Chem. Mater., 2007, 19, 6569 CrossRef CAS; (m) Q.-D. Liu, J. Lu, J. Ding, M. Day, Y. Tao, P. Barrios, J. Stupak, K. Chan, J. Li and Y. Chi, Adv. Funct. Mater., 2007, 17, 1028 CrossRef CAS.
- For excellent reviews on copper-catalyzed arylation reactions, see: (a) F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954 CrossRef CAS PubMed; (b) G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054 CrossRef CAS PubMed; (c) S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400 CrossRef CAS PubMed; (d) I. P. Beletskaya and A. V. Cheprakov, Coord. Chem. Rev., 2004, 248, 2337 CrossRef CAS PubMed.
- (a) D. S. Surry and S. L. Buchwald, Chem. Sci., 2010, 1, 13 RSC; (b) J. C. Antilla, J. M. Baskin, T. E. Barder and S. L. Buchwald, J. Org. Chem., 2004, 69, 5578 CrossRef CAS PubMed; (c) K. W. Anderson, R. E. Tundel, T. Ikawa, R. A. Altman and S. L. Buchwald, Angew. Chem., Int. Ed., 2006, 45, 6523 CrossRef CAS PubMed; (d) A. Klapars, J. C. Antilla, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2001, 123, 7727 CrossRef CAS; (e) Y.-C. Teo, F.-F. Yong, C.-Y. Poh, Y.-K. Yan and G.-L. Chua, Chem. Commun., 2009, 6258 RSC.
- (a) P. Y. S. Lam, C. G. Clark, S. Saubern, J. Adams, M. P. Winters, D. M. T. Chan and A. Combs, Tetrahedron Lett., 1998, 39, 2941 CrossRef CAS; (b) T. D. Quach and R. Batey, Org. Lett., 2003, 5, 4397 CrossRef CAS PubMed; (c) B. Sreedhar, G. T. Venkanna, K. B. S. Kumar and V. Balasubrahmanyam, Synthesis, 2008, 795 CrossRef CAS PubMed; (d) J. X. Qiao and P. Y. S. Lam, Synthesis, 2011, 829 CrossRef CAS PubMed.
- (a) P. López-Alvarado, C. Avendaño and J. C. Menéndez, Tetrahedron Lett., 1992, 33, 659 CrossRef; (b) P. López-Alvarado, C. Avendaño and J. C. Menéndez, J. Org. Chem., 1995, 60, 5678 CrossRef.
- (a) P. Petiot and A. Gagnon, Heterocycles, 2013, 88, 1615 Search PubMed; (b) P. Petiot and A. Gagnon, Eur. J. Org. Chem., 2013, 5282 CrossRef CAS; (c) A. Gagnon, V. Albert and M. Duplessis, Synlett, 2010, 2936 CrossRef CAS PubMed; (d) A. Gagnon, M. Duplessis, P. Alsabeh and F. Barabé, J. Org. Chem., 2008, 73, 3604 CrossRef CAS PubMed.
- A. Gagnon, M. St-Onge, K. Little, M. Duplessis and F. Barabé, J. Am. Chem. Soc., 2007, 129, 44 CrossRef CAS PubMed.
- C. Crifar, P. Petiot, T. Ahmad and A. Gagnon, Chem.–Eur. J., 2014, 20, 2755 CrossRef CAS PubMed.
- For reviews on organobismuth reagents, see: (a) S. Condon, C. Pichon and M. Davi, Org. Prep. Proced. Int., 2014, 46, 89 CrossRef CAS; (b) S. Shimada and M. L. N. Rao, Top. Curr. Chem., 2012, 311, 199 CrossRef CAS; (c) H. Braunschweig, P. Cogswell and K. Schwab, Coord. Chem. Rev., 2011, 255, 101 CrossRef CAS PubMed; (d) G. I. Elliott and J. P. Konopelski, Tetrahedron, 2001, 57, 5683 CrossRef CAS; (e) H. Suzuki, T. Ikegami and Y. Matano, Synthesis, 1997, 249 CrossRef CAS PubMed; (f) J.-P. Finet, Chem. Rev., 1989, 89, 1487 CrossRef CAS; (g) R. A. Abramovitch, D. H. R. Barton and J.-P. Finet, Tetrahedron, 1988, 44, 3039 CrossRef CAS; (h) D. H. R. Barton and J.-P. Finet, Pure Appl. Chem., 1987, 59, 937 CrossRef CAS; (i) L. D. Freedman and G. O. Doak, Chem. Rev., 1982, 82, 15 CrossRef CAS; (j) H. Gilman and H. L. Yale, Chem. Rev., 1942, 30, 281 CrossRef CAS.
- (a) D. H. R. Barton, J.-P. Finet and J. Khamsi, Tetrahedron Lett., 1988, 29, 1115 CrossRef CAS; (b) D. H. R. Barton, J.-C. Blazejewski, B. Charpiot, J.-P. Finet, W. B. Motherwell, M. T. B. Papoula and S. P. Stanforth, J. Chem. Soc., Perkin Trans. 1, 1985, 2667 RSC.
- D. H. R. Barton, J.-P. Finet and J. Khamsi, Tetrahedron Lett., 1987, 28, 887 CrossRef CAS.
- D. M. T. Chan, Tetrahedron Lett., 1996, 37, 9013 CrossRef CAS.
- G. A. Grasa, M. S. Viciu, J. Huang and S. P. Nolan, J. Org. Chem., 2001, 66, 7729 CrossRef CAS PubMed.
- One example of N-arylation of a pyrazole with triphenylbismuth diacetate has been reported: A. Y. Fedorov and J.-P. Finet, Tetrahedron Lett., 1999, 40, 2747 CrossRef CAS.
- A. Gagnon, M. Duplessis and L. Fader, Org. Prep. Proced. Int., 2010, 42, 1 CrossRef CAS.
- (a) P. Y. S. Lam, G. Vincent, C. G. Clark, S. Deudon and P. K. Jadhav, Tetrahedron Lett., 2001, 42, 3415 CrossRef CAS; (b) N. Joubert, E. Baslé, M. Vaultier and M. Pucheault, Tetrahedron Lett., 2010, 51, 2994 CrossRef CAS PubMed.
- (a) L. J. Mueller and M. F. Dunn, Acc. Chem. Res., 2013, 46, 2008 CrossRef CAS PubMed; (b) A. Blankfield, Int. J. Tryptophan Res., 2012, 5, 27 CrossRef CAS PubMed.
- (a) J. M. Antos and M. B. Francis, Curr. Opin. Chem. Biol., 2006, 10, 253 CrossRef CAS PubMed; (b) J. Ruiz-Rodríguez, F. Albericio and R. Lavilla, Chem.–Eur. J., 2010, 16, 1124 CrossRef PubMed.
- (a) C. J. Chapman, A. Matsuno, C. G. Frost and M. C. Willis, Chem. Commun., 2007, 3903 RSC; (b) F. Ma, X. Xie, L. Ding, J. Gao and Z. Zhang, Tetrahedron, 2011, 67, 9405 CrossRef CAS PubMed.
- While Barton and Finet reported the N-arylation of the NH2 function of O-protected glycine, phenylalanine, aspartic and glutamic acids using pentavalent bismuth reagents, they never reported the arylation of the side chains of amino acids: D. H. R. Barton, J.-P. Finet and J. Khamsi, Tetrahedron Lett., 1989, 30, 937 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 986210. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra02467b |
| This journal is © The Royal Society of Chemistry 2014 |