3D graphene aerogel-supported SnO2 nanoparticles for efficient detection of NO2
Xin Liuab,
Jiashan Cuib,
Jianbo Sun*b and
Xitian Zhang*ab
aDepartment of Physics, Harbin Institute of Technology, Harbin 150001, P. R. China
bThe Key Laboratory for Photonic and Electronic Bandgap Materials, Ministry of Education, School of Physics and Electronic Engineering, Harbin Normal University, Harbin, 150025, China. E-mail: xiaohan2298@163.com; xtzhangzhang@hotmail.com
First published on 12th May 2014
Abstract
Three-dimensional (3D) graphene aerogel-supported SnO2 (SGA) nanoparticles (NPs) are presented by a one-pot solvothermal treatment of graphene oxide in the presence of SnCl4 followed by freeze drying. The size of SnO2 nanoparticles on the graphene aerogel is as small as 5–10 nm with uniform distribution. The 3D SnO2–graphene aerogel exhibits interconnected macroporous networks which will be beneficial for gas detection. The SGA shows excellent response and selectivity towards NO2 when other common gases are present at room temperature (RT). The gas sensing responses of the resulting nanocomposites demonstrate that macroporosity in the SGA significantly improves the response and recovery time compared with the 2D SnO2–graphene nanocomposite. The sensing mechanism and the reasons for the better response are also discussed.
Introduction
Gas sensors based on nanostructured sensing elements have attracted considerable interest because nanostructured materials, such as nanocrystals,1 nanotubes,2 and nanowires,3 can be used to significantly improve the sensitivity and the response time. Tin dioxide (SnO2) is an n-type semiconducting material widely used in gas-sensing applications. Tremendous efforts have been devoted to creating highly responsive gas sensors by incorporating SnO2 nanoparticles,4 nanowires,5 and nanobelts6 into sensing platforms. But at room temperature, the SnO2 surface is expected to have a small coverage of chemisorbed oxygen, and a small change in sensor conductivity will generate when the sensor is exposed to analyte gases. However, the sensitivity is below the detection limit, as SnO2 sensors have a large resistance. Existing SnO2 sensors typically operate at temperatures over 200 °C to enhance the surface adsorption/reaction kinetics and sensitivity, which requires continuous heating of the sensor and increases the sensor cost.7 High-temperature operation is also undesirable in many situations, particularly in an explosive environment where high temperatures could trigger an explosion. To overcome the obstacle, conducting carbon matrices have been introduced to fabricate nanocomposite.8,9Graphene, a single-layer sp2 carbon lattices, has been considered as one of the most appealing carbon matrices for metal oxide particles because of its outstanding charge carrier mobility, mechanical robustness, high surface area and chemical stability.10,11 Graphene-based nanocomposites have been studied for various applications, including Li-ion batteries,12,13 catalysis,14,15 and sensors.16,17 So far, various graphene-based gas sensors have been used to detect gaseous species such as NO2 at room temperature due to the variation in the electrical resistance of graphene upon exposure to these gases.18–21 Although graphene presents excellent sensitivities to gas molecules, the performance of the sensors should be further improved to meet the requirements of practical gas sensors, e.g., high sensitivity, sensitivity and rapid response/recovery time. Recently, SnO2 and graphene composite can greatly improve the performance in terms of sensitivity or selectivity for the NO2 detection.22–24 However, the recovery process of the sensors is still slow.
More recently, it was revealed that the assembly of 2D graphene sheets into 3D architectures can provide resultant graphene-based composites with strong mechanical strength, and fast mass and electron transport kinetics due to the combination of the 3D interconnected framework and the intriguing properties of graphene. Following this theoretical work, graphene with 3D structure has been successfully synthesized via various novel methods, such as “bottom-up” molecular synthesis,25 and CVD derived structures,26 which have been utilized in supercapacitors,27 gas storage,28 and so on. Nevertheless, to the best of our knowledge, a study of SnO2 NPs supported on 3D interconnected graphene as a NO2 gas sensor has not been reported to date.
Herein, we report a facile one-step method for preparation of SGA with interconnected macroporous networks. The enhanced sensing performance of SGA toward NO2 is demonstrated at RT. The continuous porous structure and the tiny NPs which offer a large number of active sites for gas adsorption and desorption play critical roles in the fast response and recovery time. Gas detection mechanism of the as-prepared SGA product is also discussed. The as-obtained SGA exhibit a high gas-sensing activity for NO2 at RT, making it a promising candidate for practical NO2 detectors.
Experiments
Graphite oxide (GO) was synthesized from natural graphite flakes by a modified Hummers methods.29 SGA was prepared by solvothermal assembly of GO, SnCl4, sodium citrate and NaAc followed by freeze drying. In a typical experiment, 0.5 mmol SnCl4·5H2O was added to GO solution (20 mg of GO was dispersion in the 20 ml of ethylene glycol solution) to achieve the electrostatic adsorption of Sn4+ ion on GO under mechanical stirring, then 0.1 g of sodium citrate and 0.6 g of NaAc were added to form a stable complex solution. Subsequently, the mixture was transferred into a Teflon-lined stainless-steel autoclave and kept at 200 °C for 10 h. After the solvothermal procedure, the autoclave cooled naturally to RT. Finally, a black integrated SGA was obtained via a freeze-drying process to maintain the 3D monolithic architecture. The 2D graphene–SnO2 composite was synthesized in the same reaction condition. After completion of the reaction, the composite was collected by centrifugation, repeatedly washed with distilled water and absolute ethanol, and dried at 60 °C in air which was not via a freeze-drying process.The crystal structure of the as-prepared product was investigated by X-ray diffraction (XRD, D/max2600, Rigaku, Japan) with Cu Kα radiation of wavelength λ = 1.5418 Å. The morphology and microstructure were characterized by field-emission scanning electron microscopy (FE-SEM, SU70, Hitachi, Japan). Transmission electron microscopy (TEM), high-resolution transmission electron microscopy (HRTEM), and selected-area electron diffraction (SAED) measurements were obtained on a FEI Tecnai F20 microscope operated at 200 kV equipped with energy dispersive X-ray spectrometer (EDX). Raman spectra of the products were characterized by Micro-Raman spectrometer (J-Y; HR800, France) under excitation wavelength of 488 nm. Thermogravimetric analysis (TGA) was measured by a Diamond 6300. Nitrogen adsorption–desorption isotherms at 77 K were determined by NOVA 2000E.
Gas sensors were fabricated as follows: the as-prepared powder was mixed with the deionized water in order to make a paste, which was coated onto an alumina tube (4 mm in length, 1.2 mm in external diameter, and 0.8 mm in internal diameter, attached with a pair of gold electrodes) by a small brush to form a thick film. The thickness of sensing films was about 100 μm. A pair of gold electrodes was installed at each end of the ceramic tube before it was coated with the paste, and each electrode was connected with two Pt wires. The gas-sensing properties of the samples were evaluated with a self-assembly gas-sensing characterization system. The measurement was processed by a static process: a given amount of the tested gas was injected into a closed glass chamber, and the sensor was put into the chamber for the measurement of the sensitive performance. The response of the sensor was defined as S = ΔR/R0 = (R − R0)/R0 where R and R0 are the real-time and initial resistances, respectively. In addition, the response time was defined as the time required for the resistance to reach 90% of the equilibrium value after a test gas was injected, and the recovery time was the time necessary for a sensor to attain a resistance of 10% below its original value in air.
Results and discussion
The structure and morphology of as-prepared SGA were investigated by means of XRD, SEM and TEM. The representative XRD pattern confirms the formation of SnO2 (JCPDS no. 41-1445) in the composite (Fig. 1). Remarkably, no apparent diffraction peak for graphite could be identified, indicating that SnO2 NPs were efficiently deposited on the graphene surface, suppressing the restacking of graphene layers. By the incorporation of nanoparticles into the graphene sheets, the aggregation of the SnO2 NPs and restacking problems of the graphene sheets could be minimized, favoring the maintenance of a high surface area and other intrinsic chemical and physical properties of graphene.30 | ||
| Fig. 1 XRD pattern of the as-prepared SnO2–graphene aerogel. | ||
The digital photo of the resulting monolithic graphene aerogel is shown in Fig. 2a. The morphology of as-prepared SGA was characterized by means of SEM. The as prepared SGA exhibits an interconnected, porous 3D graphene framework with continuous macropores in the micrometer size range (Fig. 2b and c). Apart from the decoration of SnO2 NPs on both sides of the graphene sheets (Fig. 2d), it is noteworthy that the SnO2 NPs are distributed and tightly anchored on the graphene layers (Fig. 2d), suggesting efficient assembly between the NPs and the graphene sheets.
 | ||
| Fig. 2 Morphology of the as-synthesized sample. (a) A photograph of a graphene aerogel-supported SnO2 NPs. (b)–(d) Typical SEM images with different magnification of SGA revealing the 3D macroporous structure and uniform distribution of SnO2 NPs in the graphene aerogel. | ||
The detailed morphology and internal structure of the SGA were further studied by TEM. TEM characterization further confirms the uniform distribution of SnO2 NPs (with sizes of 5–10 nm) on the graphene (Fig. 3a and b). The HRTEM image as shown in Fig. 3c displays clearly the interplanar distances of 0.33, 0.26 and 0.24 nm consistent with the (110), (101) and (200) atomic planes of SnO2, respectively. In addition, the SAED shown in Fig. 3d of the SGA confirms the polycrystalline structure of SnO2 NPs.
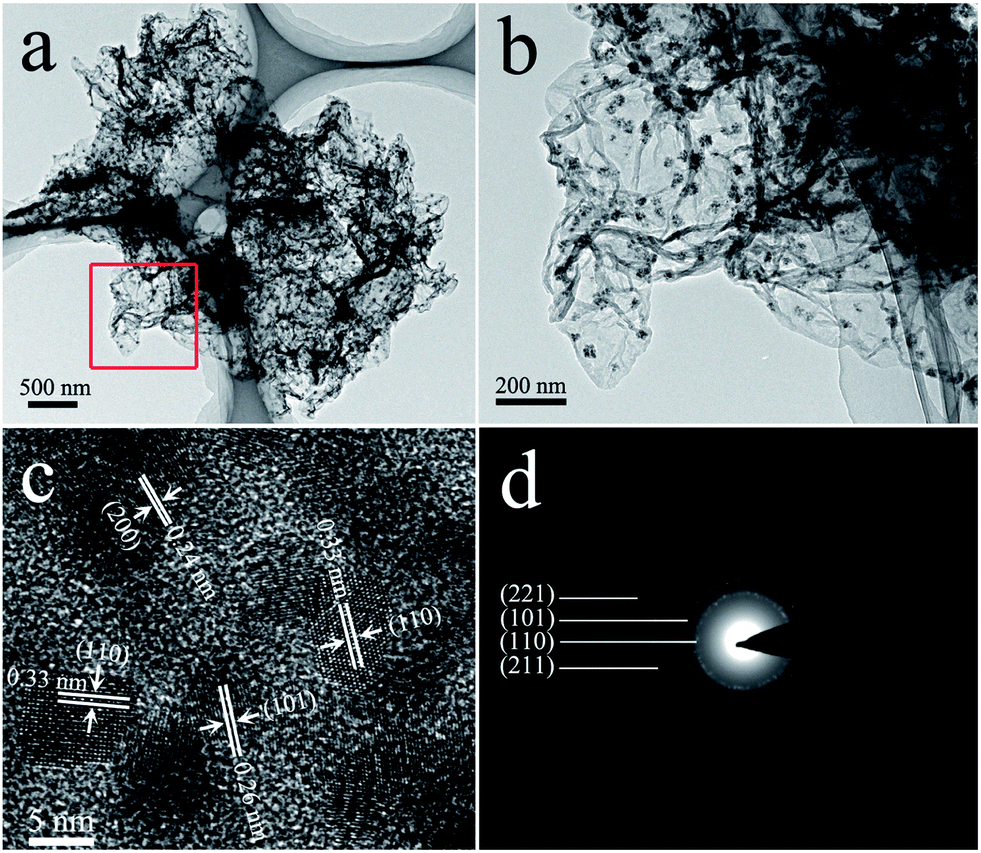 | ||
| Fig. 3 Representative (a) and (b) TEM, (c) HRTEM and (d) SAED images of SGA. | ||
Scanning TEM (STEM) and elemental mapping analysis of SGA suggest the presence of C, O, and Sn components in the composite (Fig. 4a–d). TGA measurement carried out was used to determine the chemical composition of SnO2–GA. As shown in Fig. 4e, the TGA curve displays a significant weight loss at approximately 400 °C. The miniscule weight loss that appeared below 300 °C is most likely attributed to the evaporation of adsorbed water molecules. The major weight loss from 300 to 500 °C was approximately 10%, indicating the combustion of graphene. Therefore, the content of SnO2 in SnO2–GA was calculated to be 90%. Raman spectroscopy is a forceful mean to investigate the modification of graphene and their derivatives.31,32 The Raman spectrum of SGA shown in Fig. 4f exhibits two strong peaks, corresponding to the D-band line (ca. 1342 cm−1) and the G-band line (ca. 1589 cm−1). The D-band is caused by edges or structural defects which can break the selection rule and symmetry while the G-band can be attributed to the first-order scattering of the sp2 carbon domains.31–35
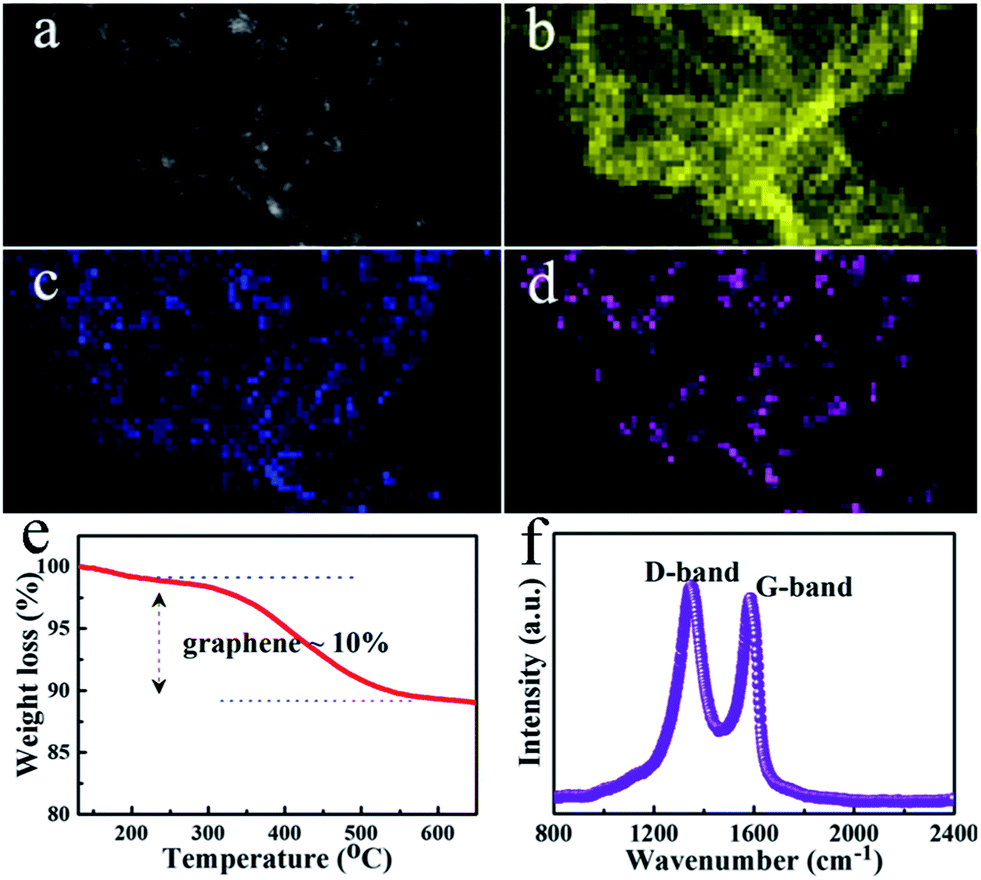 | ||
| Fig. 4 STEM, elemental mapping, and Raman analysis of SGA. (a) Typical STEM image. (b)–(d) Corresponding elemental mapping images of (b) C, (c) O, and (d) Sn. (e) TGA curve for the SGA. (f) Raman spectrum of the SGA. | ||
To further obtain the information about the as-prepared SGA, the nitrogen adsorption and desorption measurements were performed at 77 K. The representative N2 adsorption and desorption isotherm of the aerogel are shown in Fig. 5 which demonstrates the wide distribution of pore size. The specific surface area of the product was calculated to be 148.9 m2 g−1 by the Brunauer–Emmett–Teller (BET) method.
 | ||
| Fig. 5 Nitrogen adsorption–desorption isotherm of as-synthesized SGA sample. | ||
The sensing capability of the SGA was investigated systematically. Previous studies of NO2 sensing with ZnO, In2O3, and SnO2 nanostructures indicated that high sensitivities could be achieved only at elevated temperatures, e.g., ca. 200 °C.36–38 By contrast, our SGA rapidly detected NO2 gas at RT. The real-time responses of SGA were measured for various concentrations of NO2 gas (Fig. 6). When the SGA sensors were exposed to NO2 gas at RT, sensitivities and fast responses were observed; these gas sensors displayed reversible and reproducible real-time responses. Fig. 6a shows the responses upon sequential exposure as a function of analyte concentration (10, 50, 80, 120, 150 and 200 ppm). When the SGA was exposed to an electron-withdrawing gas (NO2), the resistance decreased sharply which was consistent with an increase in the number of charge carriers. Fig. 6b plots the sensitivity change of the sensors as a function of gas concentration according to the Fig. 6a which shows the increase in the responses depends near linearly on the gas concentrations in the range from 10 to 200 ppm for the sensor. Fig. 6c shows the response of SGA upon periodic exposure to 50 ppm of NO2 indicating a stable and repeatable characteristic. The response of 2D SnO2–graphene composite upon periodic exposure to 50 ppm of NO2 is also shown in Fig. 6c. Testing over three cycles, 2D counterpart revealed lower responses and sensitivity at RT. The comparative dynamic response characteristics of the sensor based on as-prepared SGA and 2D SnO2–graphene composite without freeze-drying process were shown in Fig. 6d. The as-synthesized SGA shows more rapid response and recovery time compared to the conventional SnO2–graphene composite. When exposed to 50 ppm NO2 at RT, the response/recovery time of SGA are 190 s and 224 s, respectively. However, the recovery process of the 2D SnO2–graphene composite takes several hours to completely recover to the initial state. The improved performance of the SGA should be attributed to its unique features. Compared to the 2D SnO2–graphene composite, the graphene sheets of SGA build an excellent 3D conductive network which can promote electron transfer.39 Furthermore, with the advantages of continuous porosity and high surface area, SGA gives rise to a large contact area for NO2 gas, providing fast and versatile transport pathways for gas to diffusion.
 | ||
| Fig. 6 (a) Response of the sensor to different concentrations of NO2 gas at RT. (b) Calibration sensitivity line of the sample as a function of NO2 gas. (c) Periodic exposure to NO2 gas of 50 ppm for the SGA and 2D SnO2–graphene composite at RT. (d) Response of SGA and 2D SnO2–graphene composite to 50 ppm NO2 at RT. | ||
To check the selectivity of SGA sensing material and 2D SnO2–graphene composite, we also measured the response to some typical combustible and toxic gases. Fig. 7 revealed the response of the sensor using them to various gases including CO, H2, NH3, C3H6O, CH3(CH2)3OH and CH3OH at RT. It is clear that the both sensing materials exhibit the largest response to NO2, among the tested gases. Such result indicates that the SGA exhibits a superior selectivity to NO2 against other tested gases at RT.
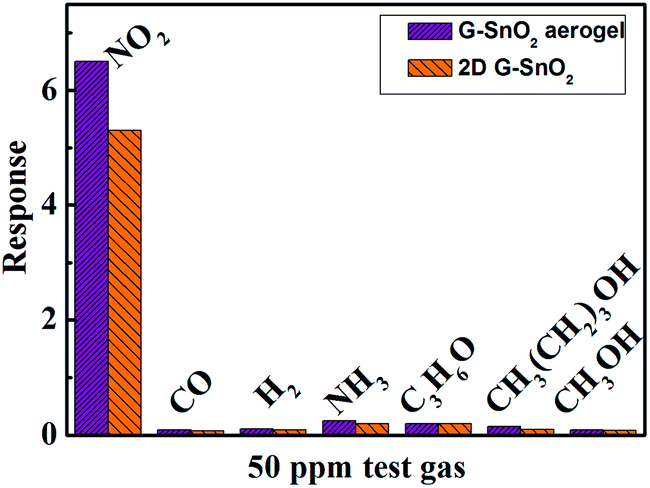 | ||
| Fig. 7 Comparison of responses of SGA and SnO2–graphene composite to various gases at RT. | ||
The sensing mechanism of the SGA is described as shown in Fig. 8. NO2 is a strong oxidizing gas (electron acceptor), so when SGA are exposed to NO2 gas, electrons are transferred from the SGA to the NO2; and finally, the NO2 transforms into NO gas and adsorbed O2−, leading to an increased number of holes in the SGA and a decrease in the electrical resistance because graphene have p-dominant conducting properties.40 In addition, at the interface between SnO2 and graphene, due to forming p–n junction and the depletion area, more electrons could flow from the graphene toward NO2 through the bonded SnO2 pathway which leads to the enhanced sensitivity. And the improvement of sensing properties compared with 2D graphene–SnO2 are mainly related to the reasons as followed: the excellent sensitivity is also attributed to the large specific surface area of the graphene aerogel which lead to highly effective surface interactions between the target gas molecules and more surface active sites. In addition, the rapid response at low temperature is due to the high porosity of SGA which could provide favorable transport pathways for gas to realize rapidly adsorption and desorption.
 | ||
| Fig. 8 NO2 gas detection mechanism of SGA. | ||
Conclusions
In summary, we have prepared 3D macroscopic SGA via a one-pot solvothermal treatment and the as-prepared SGA was explored as a novel NO2 gas sensing material. The superior sensing performance of the SGA at RT is primarily ascribed to the unique structure of the SGA. This study demonstrates an effective way to fabricate highly efficient NO2 gas transducers due to the 3D architectures which could improve recovery problems at RT. When considering the plentiful properties of both SnO2 and graphene, the SGA is promising for applications in many research fields in the future, such as supercapacitors and gas storage.Acknowledgements
This work was partially supported by the Youth academic elite funded projects of Harbin Normal University, China (no. 12XQXG30), Science and Technology Research Project of Heilongjiang Province Department of Education, China (no. 12541224) and the Natural Science Foundation of China (no. 11074060, 51172058).References
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607 CrossRef CAS PubMed.
- J. Kong, N. R. Franklin, C. W. Zhou, M. G. Chapline, S. Peng, K. J. Cho and H. J. Dai, Science, 2000, 287, 622 CrossRef CAS.
- Y. Cui, Q. Q. Wei, H. K. Park and C. M. Lieber, Science, 2001, 293, 1289 CrossRef CAS PubMed.
- B. Panchapakesan, R. Cavicchi, S. Semancik and D. L. DeVoe, Nanotechnology, 2006, 17, 415 CrossRef CAS.
- A. Kolmakov, Y. X. Zhang, G. S. Cheng and M. Moskovits, Adv. Mater., 2003, 15, 997 CrossRef CAS.
- C. Yu, Q. Hao, S. Saha, L. Shi, X. Y. Kong and Z. L. Wang, Appl. Phys. Lett., 2005, 86, 063101 CrossRef PubMed.
- W. Gopel, Sens. Actuators, A, 1996, 56, 83 CrossRef.
- X. An, J. C. Yu, Y. Wang, Y. Hu, X. Yu and G. Zhang, J. Mater. Chem., 2012, 22, 8525 RSC.
- S. Srivastava, K. Jain, V. N. Singh, S. Singh, N. Vijayan, N. Dilawar, G. Gupta and T. D. Senguttuvan, Nanotechnology, 2012, 23, 205501 CrossRef PubMed.
- D. Q. Wu, F. Zhang, H. W. Liang and X. L. Feng, Chem. Soc. Rev., 2012, 41, 6160 RSC.
- D. Q. Wu, F. Zhang, P. Liu and X. L. Feng, Chem. –Eur. J., 2011, 17, 10804 CrossRef CAS PubMed.
- B. J. Li, D. W. Rooney, N. Q. Zhang and K. N. Sun, ACS Appl. Mater. Interfaces, 2013, 5, 5057 CAS.
- W. Y. Zhang, Y. Zeng, C. Xu, H. T. Tan, W. L. Liu, J. X. Zhu, N. Xiao, H. H. Hng, J. Ma, H. E. Hoster, R. Yazami and Q. Y. Yan, RSC Adv., 2012, 2, 8508 RSC.
- Y. Q. Zhu, C. Li and C. B. Cao, RSC Adv., 2013, 3, 11860 RSC.
- I. V. Lightcap, T. H. Kosel and P. V. Kamat, Nano Lett., 2010, 10, 577 CrossRef CAS PubMed.
- J. Zhang, C. Zhao, P. A. Hu, Y. Q. Fu, Z. L. Wang, W. W. Cao, B. Yang and F. Placido, RSC Adv., 2013, 3, 22185 RSC.
- S. Mao, S. M. Cui, G. H. Lu, K. H. Yu, Z. H. Wen and J. H. Chen, J. Mater. Chem., 2012, 22, 11009 RSC.
- Z. Y. Zhang, R. J. Zou, G. S. Song, L. Yu, Z. G. Chen and J. Q. Hu, J. Mater. Chem., 2011, 21, 17360 RSC.
- M. Gautam and A. H. Jayatissa, J. Appl. Phys., 2012, 111, 094317 CrossRef PubMed.
- J. L. Johnson, A. Behnam, S. J. Pearton and A. Ural, Adv. Mater., 2010, 22, 4877 CrossRef CAS PubMed.
- Q. W. Huang, D. W. Zeng, H. Y. Li and C. S. Xie, Nanoscale, 2012, 4, 5651 RSC.
- H. Zhang, J. C. Feng, T. Fei, S. Liu and T. Zhang, Sens. Actuators, B, 2014, 190, 472 CrossRef CAS PubMed.
- S. M. Cui, Z. H. Wen, E. C. Mattson, S. Mao, J. B. Chang, M. Weinert, C. J. Hirschmugl, M. G. -Josifovskab and J. H. Chen, J. Mater. Chem. A, 2013, 1, 4462 CAS.
- S. Mao, S. M. Cui, G. H. Lu, K. H. Yu, Z. H. Wen and J. H. Chen, J. Mater. Chem., 2012, 22, 11009 RSC.
- Z. W. Xu, Z. Li, C. M. B. Holt, X. H. Tan, H. L. Wang, B. S. Amirkhiz, T. Stephenson and D. Mitlin, J. Phys. Chem. Lett., 2012, 3, 2928 CrossRef CAS.
- X. M. Li, T. S. Zhao, K. L. Wang, Y. Yang, J. Q. Wei, F. Y. Kang, D. H. Wu and H. W. Zhu, Langmuir, 2011, 27, 12164 CrossRef CAS PubMed.
- X. M. Li, T. S. Zhao, Q. Chen, P. X. Li, K. L. Wang, M. L. Zhong, J. Q. Wei, D. H. Wu, B. Q. Wei and H. W. Zhu, Phys. Chem. Chem. Phys., 2013, 15, 17752 RSC.
- G. Q. Ning, C. G. Xu, L. Mu, G. J. Chen, G. Wang, J. S. Gao, Z. J. Fan, W. Z. Qian and F. Wei, Chem. Commun., 2012, 48, 6815 RSC.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- J. F. Liang, W. Wei, D. Zhong, Q. L. Yang, L. D. Li and L. Guo, ACS Appl. Mater. Interfaces, 2012, 4, 454 CAS.
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126 CrossRef CAS PubMed.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS.
- K. N. Kudin, B. Ozbas, H. C. Schniepp and I. A. Aksay, Nano Lett., 2008, 8, 36 CrossRef CAS PubMed.
- M. S. Dresselhaus, A. Jorio, M. Hofmann, G. Dresselhaus and R. Saito, Nano Lett., 2008, 8, 2277 CrossRef PubMed.
- B. J. Li, H. Q. Cao, J. X. Zhang, M. Z. Qu, F. Lian and X. H. Kong, J. Mater. Chem., 2012, 22, 2851 RSC.
- P. Rai and Y. T. Yu, Sens. Actuators, B, 2012, 173, 58 CrossRef CAS PubMed.
- P. C. Xu, Z. X. Cheng, Q. Y. Pan, J. Q. Xu, Q. Xiang, W. J. Yu and Y. L. Chu, Sens. Actuators, B, 2008, 130, 802 CrossRef CAS PubMed.
- A. Forleo, L. Francioso, M. Epifani, S. Capone, A. M. Taurino and P. Siciliano, Thin Solid Films, 2005, 490, 68 CrossRef CAS PubMed.
- J. F Liang, Y. K. Liu, L. Guo and L. D. Li, RSC Adv., 2013, 3, 11489 RSC.
- I. Sayago, H. Santos, M. C. Horrillo, M. Alexandere, M. J. Fernandez, E. Terrado, I. Tacchini, R. Aroz, W. K. Maser, A. M. Benito, M. T. Martinez, J. Gutierrez and E. Munoz, Talanta, 2008, 77, 758 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |