
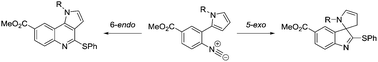
Tuneable radical cyclisations: a tin-free approach towards tricyclic and spirocyclic heterocycles via a common precursor†
Abstract
A novel common precursor approach towards both tricyclic and spirocyclic heterocycles is described. Cyclisations are based on thiyl radical/isocyanide methodology and avoid the use of tin.
 Please wait while we load your content...
Please wait while we load your content...

