NIS-mediated regioselective amidation of indole with quinazolinone and pyrimidone†
Suman Kr Ghosh and
Rajagopal Nagarajan*
School of Chemistry, University of Hyderabad, Hyderabad-500046, India. E-mail: rnsc@uohyd.ernet.in; Fax: +91-40-23012460
First published on 4th April 2014
Abstract
A mild, metal-free condition was developed for the direct regioselective C2 amidation of indoles and pyrroles with quinazolinone and pyrimidone derivatives in intermolecular fashion, which led to novel indolyl/pyrrolyl quinazolinone and pyrimidone derivatives in moderate to good yields.
Introduction
Indole1 and pyrrole2 structural motifs are considered as ‘privileged’ skeletons in numerous biologically active natural products. Hence, there is continual interest among chemists for the direct functionalization of indoles and pyrroles in regioselective fashion.3 A large number of reports by various groups have focused on forming C–C bonds regioselectively at C2 position of indole4 as well as pyrrole;5 however, only few reports are available on formation of C–N bond at the C2 position. In most cases, regioselective amination of indole has been achieved by using metal or other harsh conditions by different research groups.6 However, reports for C2 amination by using mild conditions without metal are scarce. Recently, Huang's group demonstrated C–N bond formation at indole C2 with azole in the presence of iodine.7 However, regioselective amidation of indole and pyrrole has always remained a challenge to the scientific community. Very few reports regarding amidation at C2 of indoles8 and pyrroles9 are available. Our group also reported the palladium-catalysed Buchwald cross-coupling reaction at the C2 position of indole.10 Very recently, Li's group presented direct amidation on indole by using the CDC process.11 However, to date, reports for metal-free direct amidation at indole C2 are limited. In 2008, Baran's group reported a C2 amidation in indole in an intramolecular fashion to synthesize psychotrimine.12 Very recently, Ji and coworkers also reported intramolecular amidation at the indole C2 with sulphonamides.13 Liang's group also described iodine-mediated intermolecular amidation of N-protected indoles with tosylbenzenamine.14 However, to the best of our knowledge, there is no report for metal-free direct C2 amidation of indole with cyclic amides in an intermolecular manner to date. Numerous biologically active natural products contain C–N amide linkage at the C2 position of indole, such as asperazine and chetomin (Fig. 1). Most of these natural products contain cyclic amide linkage, but direct amidation with cyclic amides at the C2 position of indole still remains unexplored. | ||
| Fig. 1 Naturally occurring indole and pyrrole alkaloids containing C–N amide linkage. | ||
Quinazolinone15 and pyrimidones16 are very important classes of heterocycles, due to their diverse range of biological properties like anticancer, anti-inflammatory, diuretic, anticonvulsant, and antihypertensive properties. Studies have shown that the functionalization of quinazolinone's amide N–H with alkyl, aryl groups increases the activity of quinazolinone motif.17 However, heterocycles like indoles, pyrroles and other cyclic amides have not been explored elaborately as a functionality to date.18 Thus, indolylquinazolinone product may possess some novel biological activity, which can be further explored. Therefore, in this letter, we wish to report a novel N-iodosuccinimide-mediated protocol for direct C2 amidation of indoles and pyrroles with cyclic amides like pyrimidone/quinazolinone derivatives.
Results and discussion
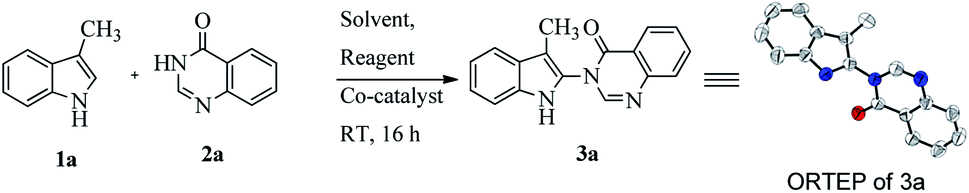
To develop a metal-free condition for selective amidation in an indole moiety, we began our study with the reaction of 3-methylindole (1a; 1.1 equiv.) and quinazolinone (2a; 1.0 equiv.) with an iodination source such as N-iodosuccinimide (NIS; 1.2 equiv.) in CH3CN solvent at room temperature. After 16 h, we were delighted to find the desired product 3-(3-methyl-1H-indol-2-yl)-3H-quinazolin-4-one (3a) in 30% yield. The relative structure of compound 3a was determined by detailed spectroscopic analysis and X-ray crystallographic study (Table 1). Next, to improve the yield of the desired product 3a, we screened a variety of both polar and nonpolar solvents (entries 1–12). Polar solvents such as CH3CN, THF, DMSO and EtOAc (entries 1–4) gave only moderate yields of the desired product. Interestingly, when CHCl3 was used as the solvent, which is relatively nonpolar in nature, the yield of 3a was 70% (entry 5).
|
||||
|---|---|---|---|---|
| Entry | Solvent | Reagent/catalyst | Co-catalystb | Yieldsc 3a [%] |
| a Unless otherwise specified, reaction was performed on 0.34 mmol scale with 1a (1.1 equiv.), 2a (1.0 equiv.), reagent (1.2 equiv.) and solvent (5 ml) at RT. The reaction time was 16 h.b Co-catalyst used 10 mol%.c Isolated yields.d NIS used was 2 equiv.e NIS used was 30 mol%. | ||||
| 1 | CH3CN | NIS | — | 30 |
| 2 | THF | NIS | — | 10 |
| 3 | DMSO | NIS | — | 40 |
| 4 | EtOAc | NIS | — | 58 |
| 5 | CHCl3 | NIS | — | 70 |
| 6 | Toluene | NIS | — | 15 |
| 7 | Benzene | NIS | — | 34 |
| 8 | p-Xylene | NIS | — | 20 |
| 9 | DCM | NIS | — | 53 |
| 10 | DCE | NIS | — | 64 |
| 11 | 1,2-DCB | NIS | — | 42 |
| 12 | H2O | NIS | — | 40 |
| 13 | Neat | NIS | — | 33 |
| 14 | CHCl3 | NBS | — | 62 |
| 15 | CHCl3 | I2 | — | 67 |
| 16 | CHCl3 | NIS | CuI | 62 |
| 17 | CHCl3 | NIS | CuBr | 56 |
| 18 | CHCl3 | NIS | DIB | 20 |
| 19 | CHCl3 | NIS | KI | 30 |
| 20d | CHCl3 | NIS | — | 70 |
| 21e | CHCl3 | NIS | — | 20 |
As a result, we used nonpolar solvents like toluene, benzene, p-xylene, but unfortunately they were inferior to CHCl3 in terms of yields (entries 6–8). Therefore, we assumed that chlorinated solvents might give a better yield compared with CHCl3. Thus, we used some chlorine-containing solvents like DCM, DCE and 1,2-dichlorobenzene (entries 9–11); however, among them, only DCE was able to give a maximum yield of 64%. Moreover, using water and neat-reaction conditions also failed to improve the yield (entries 12–13). Thus, considering CHCl3 as the optimized solvent, we varied other parameters. When N-bromosuccinimide (entry 14) was used instead of NIS, the yield decreased, whereas in the case of iodine (entry 15), the yield remained almost unaffected. Furthermore, the screening of co-catalysts like CuI, CuBr, DIB, KI in 10 mol%, led to no improvement of the reaction performance.
To verify the role of NIS, i.e., whether it was catalytic or stoichiometric, we carried out two reactions, in which NIS had been used in 2 equiv. (entry 20) and 30 mol% (entry 21). The use of catalytic amounts of NIS decreased the yield of the product, whereas higher loading of NIS failed to furnish more than 70% of the desired product.
From the summarized results in Table 1, the reaction of 1.0 equiv. of quinazolinone 2a, with 1.1 equiv. of indole 1a, using 1.2 equiv. of NIS in CHCl3 solvent at RT for 16 h was deemed to be the optimum condition (entry 5). With these optimized conditions in hand, the generality and scope of the reaction was explored for a range of 3-substituted indole, as well as 1,3 disubstituted indole, with quinazolinone derivatives and pyrimidone (Scheme 1). A variety of functional groups such as moderate electron-withdrawing and electron-releasing groups in substituted indoles were well tolerated to give moderate to good yield of indolylquinazolinone and indolylpyrimidone products (3a–n). When electron-donating substituents at indole C3 were coupled with quinazolinones and pyrimidones, a good yield of the corresponding products was obtained (3a,d). However, 6-bromoquinazolinone and benzoquinazolinone, which are electron deficient, gave a lower yield of the product (3b–c), whereas when methyl was substituted with a moderate electron-withdrawing group like CH2CO2Me, the yields of the corresponding products remained almost unaffected (3f–g).
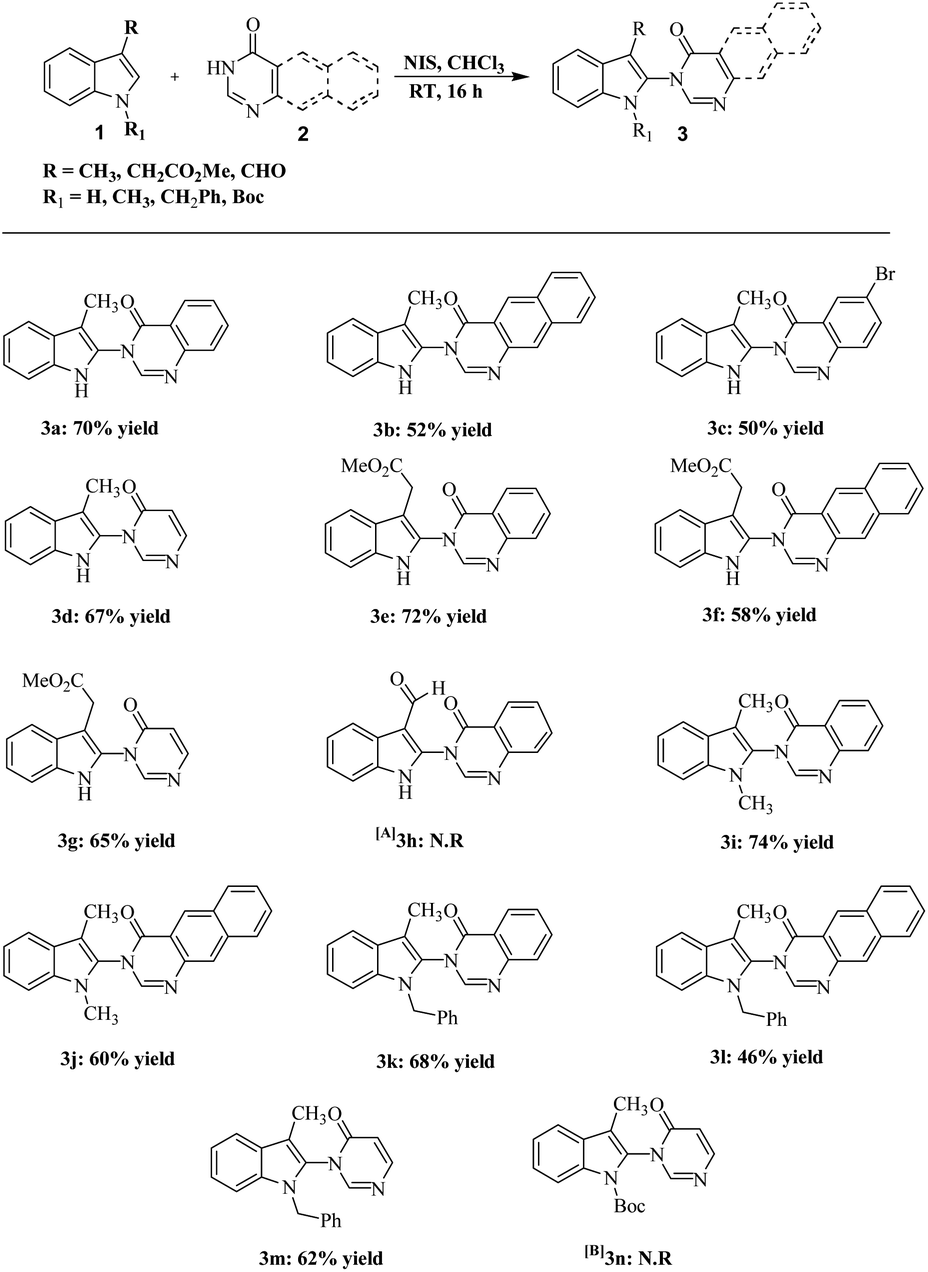 | ||
| Scheme 1 Synthesis of indolylquinazolinone derivatives. Unless otherwise specified, the reaction was performed on 0.68 mmol scale with 1 (1.1 equiv.), 2 (1 equiv.), NIS (1.2 equiv.) in solvent (7 ml) at RT. [A] Reaction time was 72 h. [B] Reaction time was 48 h. N.R. = no reaction. | ||
Surprisingly, when the aldehyde group was placed at C3 of indoles, the reaction did not proceed even after three days (3h). After examining electron-donating as well as electron-withdrawing groups in C3 position of indoles, we tested the feasibility of the reaction with 1,3-disubstituted indoles. We found that substituent like methyl and benzyl groups on nitrogen were well tolerated (3i–m). It is worth mentioning that 1,3-dimethylindole gave a better yield with the corresponding indolylquinazolinone product compared with the 3-methylindole. When the indole nitrogen was protected with an electron-withdrawing group, Boc, the reaction did not proceed even after 48 h (3n). This may be due to the lowering of nucleophilicity on the indole nitrogen, which plays a vital role in iodination. Subsequently, we expanded the scope of the reaction with the C3 unsubstituted indoles and applied our optimized condition on plain indoles and quinazolinones. The outcome of this reaction, which gave the expected product 5a, was not surprising; however, along with the product a lower percentage of 3-iodoindolylquinazolinone (5aa) was also obtained. We suspected that the variation in the amount of NIS may give one of the products specifically. Thus, we took indoles and quinazolinones as the coupling products and used only 1.05 equiv. of NIS instead of 1.2 equiv. under the same optimized conditions. After 16 h, we obtained 3-(1H-indol-2-yl)-3H-quinazolin-4-one (5a), whereas when 1.7 equiv. of the NIS was used, the reaction gave only 3-(3-iodo-1H-indol-2-yl)-3H-quinazolin-4-one (5aa) specifically (Fig. 2).
Furthermore, this reaction also tolerated 5- and 6-bromoindoles and 5-methoxyindole, as well as afforded the targeted product along with the undesired iodo-substituted product. The yields of both products depended on the amount of the NIS and the time of the reaction. The 1H NMR clearly showed the variation in yield between the desired indolylquinazolinone and the undesired 3-iodoindolylquinazolinone products (see ESI†). Surprisingly, when 1-methylindole and 1-butylindoles reacted with quinazolinone, we obtained only our desired products (5f–g), whereas 1-sulfonylindole and 7-azaindole were inactive in this procedure. The reason behind this observation may be again justified by the lowering and increasing of the nucleophilicity of the indole nitrogen, depending upon the electron-withdrawing/releasing group (Scheme 2).
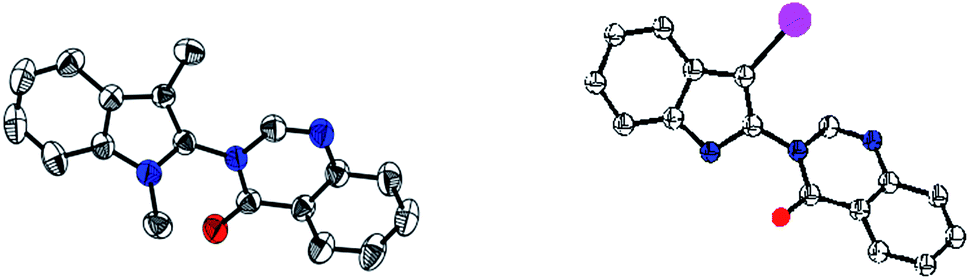 | ||
| Fig. 2 ORTEP of Compound 3i, 5aa (hydrogens are removed for clarity). | ||
After a successful encounter with indoles, we further extended the scope to pyrrole heterocycle. Because reports for regioselective C–N bond formation at the pyrrole C2 were limited, we executed our optimized condition over the pyrrole and quinazolinone moiety. Unfortunately, the reaction failed to give the desired pyrrolylquinazolinone product after 48 h of stirring at room temperature. Next, we replaced NIS with granular iodine in our optimized condition. To our delight, we obtained 38% of the desired product after 12 h.
 | ||
| Scheme 2 Synthesis of 3-unsubstituted indolylquinazolinone derivatives. Unless otherwise specified, the reaction was performed on 0.68 mmol scale with 4 (1.1 equiv.), 2a (1 equiv.), NIS (1.2 equiv.) in solvent (7 ml) at RT. [A] 1.05 equiv. of NIS was used, and reaction time 16 h. [B] 1.7 equiv. of NIS was used, and reaction time was 24 h. N.R. = no reaction. | ||
Hence, we again sought for an optimum condition with iodine and different solvents, and it was determined that iodine is the key component for this reaction to occur. The optimized conditions (1.1 equiv. of 6, 1.3 equiv. of I2, CHCl3, RT, 18 h) were found to be applicable over a range of N-substituted/unsubstituted pyrrole and quinazolinone derivatives (Scheme 3). N-substituted/unsubstituted pyrroles reacted smoothly with quinazolinone derivatives to give corresponding pyrroloquinazolinone products (7a–d) in moderate yields (Scheme 3).
 | ||
| Scheme 3 Synthesis of pyrrolylquinazolinone derivatives. Reaction was performed on 0.68 mmol scale with 6 (1.1 equiv.), 2 (1 equiv.), I2 (1.3 equiv.) in solvent (7 ml) at RT. | ||
Furthermore, we were eager to see the outcome of the reaction when aliphatic amides/imides were employed as a coupling partner with 3-methylindole. Hence, we took 3-methylindole (1.1 equiv.), benzamide (1.0 equiv.), and NBS (1.1 equiv.) at room temperature in CHCl3, and an unexpected product, 3-methyl-1,3-dihydro-indol-2-one (8), was formed in 50% yield after 4 h (Scheme 4[A]). Next, 3-methylindole (1.1 equiv.) was treated with benzanilide (1.0 equiv.) and NIS (1.1 equiv.) in CHCl3. Again, we obtained another undesired product, 3,3′-dimethyl-3′H-[1,2′]biindolyl-3′-ol (9), in 22% yield after 24 h. This unexpected compound (9) was fully characterized with NMR spectral data along with the X-ray crystallographic study. Synthesis of this kind of dimer is already reported in the literature using Co-salen complex in the oxygen atmosphere.19 It may be that the indole nitrogen is more nucleophilic, in comparison with the nitrogen of benzanilide, which led to the formation of the unexpected product (9). Thus, we suspected that protection of the indole nitrogen would be able to furnish our expected coupled product. Therefore, we used the same reaction condition with 1,3-dimethylindole. However, this time we obtained a coupled product, but instead of benzanilide, succinimide coupled to the 1,3-dimethylindole (Scheme 4[C]). The lower yield for the formation of 1-(1,3-dimethyl-1H-indol-2-yl)-pyrrolidine-2,5-dione (10) can be justified by the in situ formation of succinimide. Next, when succinimide was used as a starting material, the yield of the coupled product (10) increased to 62% (Scheme 4[D]); however, the use of phthalimide as a starting material also furnished only a succinimide-coupled product (10) (Scheme 4[E]). This may be due to the iminol form, rather than the amide form, which is participating in the reaction as shown in mechanism (Scheme 5).
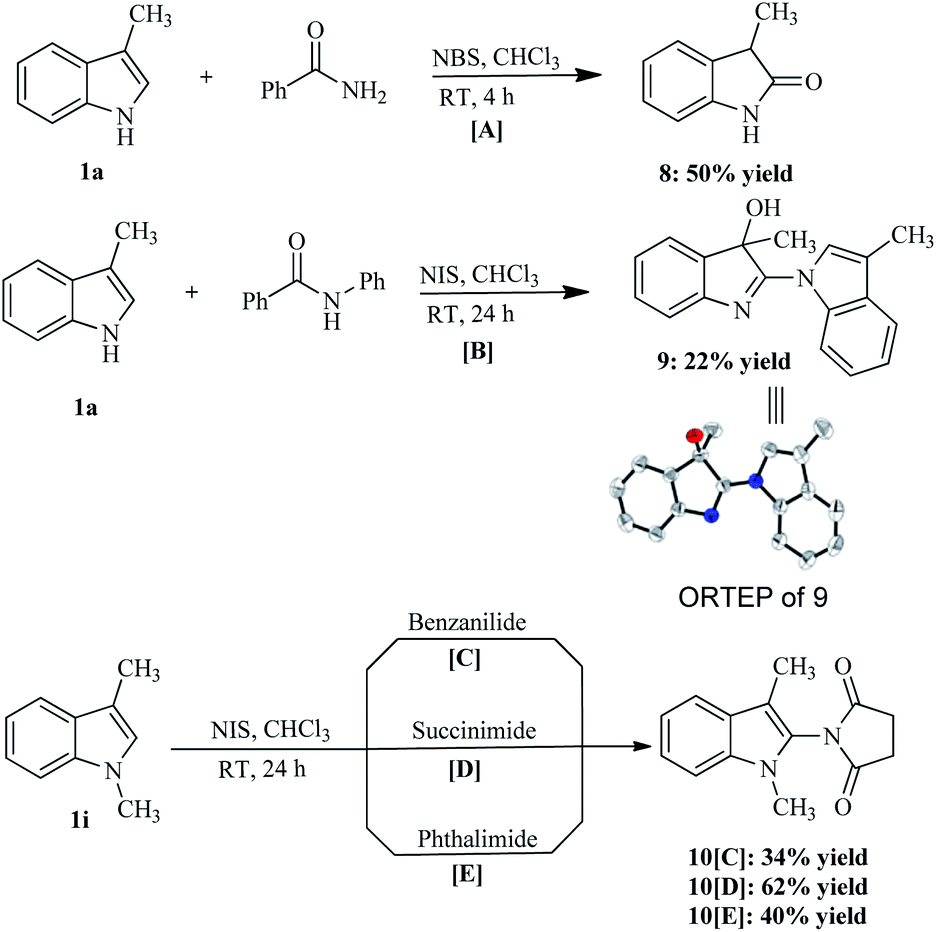 | ||
| Scheme 4 Reaction between indoles and aliphatic amides/imides. [A] 1a (0.98 mmol), benzamide (0.82 mmol) and NBS (0.98 mmol) in a solvent at RT for 4 h. [B] 1a (0.51 mmol), benzanilide (0.51 mmol), NIS (0.61 mmol) in a solvent at RT for 24 h. [C] 1i (0.35 mmol), benzanilide (0.35 mmol), NIS (0.35 mmol) in a solvent at RT for 24 h. [D] 1i (1 mmol), succinimide (1 mmol), NIS (1 mmol) in a solvent at RT for 24 h. [E] 1i (0.68 mmol), phthalimide (0.68 mmol), NIS (0.82 mmol) in a solvent at RT for 24 h. | ||
 | ||
| Scheme 5 Possible mechanism. | ||
After all of these extensive studies with a variety of substrates, and from the outcomes we proposed a possible pathway of the reaction as shown in Scheme 5. Iodination on C3 of indole with NIS produces the intermediate 11, which undergoes an immediate nucleophilic substitution with the iminol form (12) to generate the intermediate 13. Then, subsequent elimination of HI led to the expected product (14) (Scheme 5).
 | ||
| Scheme 6 Synthesis of the indolo [1,3]-diazepine skeleton. | ||
In the final part of our study, we synthesised an indolo [1,3]-diazepine skeleton fused with quinazolinone. Diazepines are a very important class of heterocycles having considerable applications in pharmaceutical industry. However, among them, 1,4-diazepine has the maximum applications, whereas, 1,3-diazepine systems are very rarely known because of their biological activity. Some 1,3-diazepine-fused heterocycles show anticancer and anti-AIDS activity as well as inhibition of HIV protease.20 Thus, we synthesised a 1,3-diazpine containing a novel indolylquinazolinone heterocycle, which may possess some interesting biological properties. We took compound 3a and treated with 1,3-dibromopropane in the presence of K2CO3 and DMF solvent at room temperature, which led to the formation of 3-[1-(3-bromo-propyl)-3-methyl-1H-indol-2-yl]-3H-quinazolin-4-one (15). Next, the treatment of compound 15 with LDA in presence of HMPA at −78 °C furnished the desired macrocyclic compound 16 in 42% yields (Scheme 6).
Conclusions
In summary, we have developed an efficient, metal-free methodology for direct amidation regioselectively at C2 in indoles and pyrroles with quinazolinones and pyrimidones. A series of novel indolylquniazolinones/pyrimidones and pyrrolylquinazolinones were prepared with free or protected indoles and pyrroles. Further, we prepared a highly functionalized 1,3-diazepine compound, which may have useful biological properties.Experimental
General experimental procedure for preparation of indolylquinazolinone
An oven dried Schlenk tube was charged with indole or its derivative (1, 0.74 mmol), quinazolinone derivatives (2, 0.68 mmol), NIS (0.81 mmol) and distilled CHCl3 (7 ml). The Schlenk tube was then flushed with nitrogen. The reaction mixture was stirred at RT for 16 h. It was then diluted with water, and the aqueous phase was extracted with DCM (30 ml). The combined organic layer was dried over Na2SO4 and concentrated using a rotary evaporator under reduced pressure. The resulting residue was purified by column chromatography on silica gel (ethyl acetate–hexane = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7) to afford the desired product.
:7) to afford the desired product.
3-(3-Iodo-5-methoxy-1H-indol-2-yl)-3H-quinazolin-4-one (5bb). Compounds were obtained as bright yellow solid mixture with ratio of (5b
:5bb = 1:0.41); (overall 108 mg, 45%); IR (KBr) 3358, 3260, 2991, 2926, 1682, 1654, 1254, 1210, 767, 690 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 12.20 (0.396H, s), 11.62 (1.00H, s), 8.46 (1.040H, s), 8.38 (0.443H, s), 8.25–8.23 (1.428H, m), 7.97–7.89 (1.433H, m), 7.82–7.76 (1.390H, m), 7.68–7.61 (1.392H, m), 7.39–7.32 (1.459H, m), 7.102 (1.063H, s), 6.94–6.92 (0.446H, m), 6.84–6.82 (1.387H, m), 6.59 (0.978H, s), 3.81 (1.184H, s), 3.76 (2.961H, s); 13C NMR (100 MHz, DMSO-d6) δ 160.3, 155.0, 154.2, 147.7, 147.4, 135.6, 132.1, 129.9, 128.4, 127.9, 127.4, 126.9, 121.9, 113.0, 112.9, 102.5, 97.9, 55.8; HRMS (ESI-MS) calcd for 5b C17H13N3O2 (M + H) 292.1086, found 292.1079; for 5bb C17H12IN3O2 (M + H) 418.0052, found 418.0051.
3-(5-Bromo-3-iodo-1H-indol-2-yl)-3H-quinazolin-4-one (5cc). Compound was obtained as bright yellow solid (111 mg, 35%); m.p. = 218 °C; IR (KBr) 3407, 2932, 1675, 1601, 1455, 1008, 938 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 12.59 (1H, s), 8.43 (1H, s), 8.26 (1H, dd, J = 8.0 Hz, J = 1.0 Hz), 7.99–7.95 (1H, m), 7.82 (1H, d, J = 8.0 Hz), 7.68 (1H, t, J = 8.0 Hz), 7.55 (1H, d, J = 1.5 Hz), 7.49 (1H, d, J = 9.0 Hz), 7.43 (1H, dd, J = 8.5 Hz, J = 2.0 Hz); 13C NMR (100 MHz, DMSO-d6) δ 160.1, 147.8, 147.5, 136.1, 134.5, 134.1, 131.1, 128.7, 128.2, 127.1, 126.9, 123.2, 121.7, 115.0, 113.6, 58.1; HRMS (ESI-MS) calcd for C16H9Br79IN3O (M + H) 465.9052, found 465.9049, C16H9Br81IN3O (M + H) 467.9031, found 467.9028.
3-(6-Bromo-3-iodo-1H-indol-2-yl)-3H-quinazolin-4-one (5dd). Compounds were obtained as bright yellow solid mixture with ratio of (5d
:5dd = 0.3:1); (overall 160 mg, 54%); IR (KBr) 3342, 2915, 1676, 1610, 1413, 1260, 772, 701 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 12.48 (1H, s), 11.96 (0.304H, s), 8.49 (0.326H, s), 8.41 (0.994H, s), 8.26–8.24 (1.35H, m), 7.98–7.90 (1.48H, m), 7.82–7.76 (1.356H, m), 7.72 (1.048H, s), 7.68–7.62 (1.672H, m), 7.59–7.57 (0.334H, m), 7.35 (2.14H, s), 7.22–7.20 (0.346H, m), 6.73 (0.298H, s); 13C NMR (100 MHz, DMSO-d6) δ 160.1, 147.9, 147.5, 147.2, 136.0, 135.9, 135.7, 134.0, 128.6, 128.4, 128.2, 127.9, 127.0, 125.9, 124.2, 123.0, 121.8, 116.8, 115.3, 114.6, 98.2, 59.1; HRMS (ESI-MS) calcd for 5d C16H10Br79N3O (M + H) 340.0085, found 340.0075; C16H10Br81N3O (M + H) 342.0065, found 342.0056, for 5dd C16H9Br79IN3O (M + H) 465.9052, found 465.9049; C16H9Br81IN3O (M + H) 467.9031, found 467.9029.
:hexane = 1:9) to afford the desired product (15) (78 mg; 55% yield). This compound was obtained as yellow sticky semisolid with little inseparable impurities; IR (KBr) 3052, 2920, 1693, 1610, 1276, 920 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.42 (1H, d, J = 7.5 Hz), 8.025 (1H, s), 7.89–7.84 (2H, m), 7.67 (1H, d, J = 8.0 Hz), 7.63 (1H, t, J = 7.5 Hz), 7.46 (1H, d, J = 8.0 Hz), 7.38–7.35 (1H, m), 7.24 (1H, t, J = 7.5 Hz), 4.24–4.10 (2H, m), 3.39–3.29 (2H, m), 2.33–2.23 (2H, m), 2.24 (3H, s); 13C NMR (125 MHz, CDCl3) δ 161.1, 147.9, 146.6, 135.1, 133.2, 128.1, 127.9, 127.4, 126.9, 123.5, 122.2, 120.0, 119.8, 117.0, 109.8, 108.5, 41.4, 32.4, 30.5, 8.1; HRMS (ESI-MS) calcd for C20H18Br79N3O (M + H) 396.0711, found 396.0711; C20H18Br81N3O (M + H) 398.0691, found 398.0693.:hexane = 3:7) to afford the desired product 16 (55 mg, 42% yield). This compound was obtained as yellow solid; m.p. = 158 °C; IR (KBr) 3046, 2958, 2920, 1693, 1605, 1457, 1260, 739 cm−1; 1H NMR (400 MHz, CDCl3 + DMSO-d6) δ 8.22 (1H, d, J = 8.0 Hz), 7.89 (1H, t, J = 7.2 Hz), 7.72 (1H, d, J = 8.0 Hz), 7.63–7.56 (3H, m), 7.23 (1H, t, J = 7.6 Hz), 7.11 (1H, t, J = 7.2 Hz), 4.62–4.57 (1H, m), 4.02–3.95 (1H, m), 2.78–2.74 (1H, m), 2.25–2.17 (3H, m), 2.11 (3H, s); 13C NMR (100 MHz, CDCl3 + DMSO-d6) δ 159.6, 156.1, 147.4, 135.4, 133.5, 127.9, 127.6, 127.6, 127.3, 127.0, 122.7, 121.0, 119.8, 119.4, 110.0, 106.2, 39.0, 32.9, 27.6, 9.4; HRMS (ESI-MS) calcd for C20H17N3O (M + H) 316.1450, found 316.1448.Acknowledgements
We are grateful to DST for financial support (project number: SR/S1/OC-70/2008) and for the single-crystal X-ray diffractometer facility in our school. S.K.G also thanks UGC for the Junior Research Fellowship.Notes and references
- For few recent reports on indole alkaloids, see: (a) H. D. H. Showalter, J. Nat. Prod., 2013, 76, 455–467 CrossRef CAS PubMed; (b) M. Chen, C. L. Shao, X. M. Fu, R. F. Xu, J. J. Zheng, D. L. Zhao, Z. G. She and C. Y. Wang, J. Nat. Prod., 2013, 76, 547–553 CrossRef CAS PubMed; (c) K. Imada, E. Sakai, H. Kato, T. Kawabata, S. Yoshinaga, T. Nehira, H. Terasawa and S. Tsukamoto, Tetrahedron, 2013, 69, 7051–7055 CrossRef CAS PubMed; (d) M. Ishikura, T. Abe, T. Choshi and S. Hibino, Nat. Prod. Rep., 2013, 30, 694–752 RSC.
- For few recent reports on pyrrole alkaloids, see: (a) J. F. Hu, H. Fan, J. Xiong and S. B. Wu, Chem. Rev., 2011, 111, 5465–5491 CrossRef CAS PubMed; (b) A. A. Mourabit, M. A. Zancanella, S. Tilvi and D. Romo, Nat. Prod. Rep., 2011, 28, 1229–1260 RSC; (c) H. Fan, J. Peng, M. T. Hamann and J. F. Hu, Chem. Soc. Rev., 2008, 108, 264–287 CrossRef CAS PubMed.
- (a) M. Shiri, Chem. Rev., 2012, 112, 3508–3549 CrossRef CAS PubMed; (b) M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608–9644 CrossRef CAS PubMed; (c) S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873–2920 CrossRef CAS PubMed.
- (a) B. Li, J. Ma, W. Xie, H. Song, S. Xu and B. Wang, Chem.–Eur. J., 2013, 19, 11863–11868 CrossRef CAS PubMed; (b) L. Jiao, E. Herdtweck and T. Bach, J. Am. Chem. Soc., 2012, 134, 14563–14572 CrossRef CAS PubMed.
- (a) L. Jiao and T. Bach, Angew. Chem., Int. Ed., 2013, 52, 6080–6083 CrossRef CAS PubMed; (b) Y. Xu, L. Zhao, Y. Li and H. Doucet, Adv. Synth. Catal., 2013, 355, 1423–1432 CrossRef CAS; (c) E. M. Beck, N. P. Grimster, R. Hatley and M. J. Gaunt, J. Am. Chem. Soc., 2006, 128, 2528–2529 CrossRef CAS PubMed.
- (a) X. Y. Liu, P. Gao, Y. W. Shen and Y. M. Liang, Org. Lett., 2011, 13, 4196–4199 CrossRef CAS PubMed; (b) M. Poirier, S. Goudreau, J. Poulin, J. Savoie and P. L. Beaulieu, Org. Lett., 2010, 12, 2334–2337 CrossRef CAS PubMed; (c) J. R. Harrison and C. J. Moody, Tetrahedron Lett., 2003, 44, 5189–5191 CrossRef CAS.
- W. B. Wu and J. M. Huang, Org. Lett., 2012, 14, 5832–5835 CrossRef CAS PubMed.
- (a) J. Shi, B. Zhou, Y. Yang and Y. Li, Org. Biomol. Chem., 2012, 10, 8953–8955 RSC; (b) J. E. Mangette, X. Chen, R. Krishnamoorthy, A. S. Vellekoop, A. J. Csaki, F. Camara, W. D. Paquette, H. J. Wang, H. Takahashi, R. Fleck and G. P. Roth, Tetrahedron Lett., 2011, 52, 1292–1295 CrossRef CAS PubMed.
- (a) J. Wu, W. Vetter, G. W. Gribble, J. S. Schneekloth, Jr, D. H. Blank and H. Gorls, Angew. Chem., Int. Ed., 2002, 41, 1740–1743 CrossRef CAS; (b) M. D. Rosa and G. C. Nieto, Tetrahedron Lett., 1988, 29, 2405–2408 CrossRef.
- (a) A. S. Kumar, P. V. A. Rao and R. Nagarajan, Org. Biomol. Chem., 2012, 10, 5084–5093 RSC; (b) A. S. Kumar and R. Nagarajan, Org. Lett., 2011, 13, 1398–1401 CrossRef CAS PubMed.
- Q. Shuai, G. Deng, Z. Chua, D. S. Bhole and C. J. Li, Adv. Synth. Catal., 2010, 352, 632–636 CrossRef CAS.
- T. Newhouse and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 10886–10887 CrossRef CAS PubMed.
- Z.-J. Cai, S.-Y. Wang and S.-J. Ji, Org. Lett., 2013, 15, 5226–5229 CrossRef CAS PubMed.
- Y. X. Li, H. X. Wang, S. Ali, X. F. Xia and Y. M. Liang, Chem. Commun., 2012, 48, 2343–2345 RSC.
- (a) M. Sharma, K. Chauhan, R. Shivahare, P. Vishwakarma, M. K. Suthar, A. Sharma, S. Gupta, J. K. Saxena, J. Lal, P. Chandra, B. Kumar and P. M. S. Chauhan, J. Med. Chem., 2013, 56, 4374–4392 CrossRef CAS PubMed; (b) S. B. Mhaske and N. P. Argade, Tetrahedron, 2006, 62, 9787–9826 CrossRef CAS PubMed.
- (a) K. J. Filipski, A. G. Perez, J. Bian, C. Perreault, G. E. Aspnes, M. T. Didiuk, R. L. Dowa, R. F. Hank, C. S. Jones, R. J. Maguire, M. Tu, D. Zeng, S. Liu, J. D. Knafels, J. Litchfield, K. Atkinson, D. R. Derksen, F. Bourbonais, K. S. Gajiwala, M. Hickey, T. O. Johnson, P. S. Humphries and J. A. Pfefferkorn, Bioorg. Med. Chem. Lett., 2013, 23, 4571–4578 CrossRef CAS PubMed; (b) I. M. Lagoja, Chem. Biodiversity, 2005, 2, 1–50 CrossRef CAS PubMed.
- (a) F. Rorsch, E. Buscato, K. Deckmann, G. Schneider, M. S. Zsilavecz, G. Geisslinger, E. Proschak and S. Grosch, J. Med. Chem., 2012, 55, 3792–3803 CrossRef PubMed; (b) T. D. Cushing, D. P. Metz, D. A. Whittington and L. R. McGee, J. Med. Chem., 2012, 55, 8559–8581 CrossRef CAS PubMed; (c) R. Karuturi, R. A. A. Horani, S. C. Mehta, D. Gailani and U. R. Desai, J. Med. Chem., 2013, 56, 2415–2428 CrossRef CAS PubMed.
- A. Kumar, S. Sharma, Archana, K. Bajaj, S. Sharma, H. Panwar, T. Singh and V. K. Srivastava, Bioorg. Med. Chem., 2003, 11, 5293–5299 CrossRef CAS.
- T. Newhouse, C. A. Lewis, K. J. Eastman and P. S. Baran, J. Am. Chem. Soc., 2010, 132, 7119–7137 CrossRef CAS PubMed , and references therein.
- (a) M. Xie, R. K. Ujjinamatada, M. Sadowska, R. G. Lapidus, M. J. Edelman and R. S. Hosmane, Bioorg. Med. Chem. Lett., 2010, 20, 4386–4389 CrossRef CAS PubMed; (b) A. Reisinger, R. Koch, P. V. Bernhardt and C. Wentrup, Org. Biomol. Chem., 2004, 2, 1227–1238 RSC; (c) P. Y. S. Lam, P. K. Jadhav, C. J. Eyermann, C. N. Hodge, Y. Ru, L. T. Bacheler, J. L. Meek, M. J. Otto, M. M. Rayner, Y. N. Wong, C. H. Chang, P. C. Weber, D. A. Jackson, T. R. Sharpe and S. E. Viitanen, Science, 1994, 263, 380–384 CAS , and references therein.
- R. G. Alvarez, I. S. Hunter, C. J. Suckling, M. Thomas and U. Vitinius, Tetrahedron, 2001, 57, 8581–8587 CrossRef CAS.
- Molecular formula: C17H13N3O1, unit cell parameters: a = 10.830(3), b = 12.205(3), c = 12.209(3) Å, α = 102.677(4)°, β = 106.686(4)°, γ = 110.923(4)° and space group P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Molecular formula: C18H15N3O1, unit cell parameters: a = 8.3989(18), b = 12.9656(14), c = 13.6993(18) Å, β = 101.702(16)° and space group P21/n. Molecular formula: C16H10I1N3O1, unit cell parameters: a = 13.0741(18), b = 4.2988(4), c = 24.974(3) Å, β = 100.040(11)° and space group P21/n. Molecular formula: C18H16N2O1, unit cell parameters: a = 10.8278(7), b = 11.3669(8), c = 13.0687(6)Å, α = 100.716(5)°, β = 106.227(5)°, γ = 104.477(6)° and space group P.†.
. Molecular formula: C18H15N3O1, unit cell parameters: a = 8.3989(18), b = 12.9656(14), c = 13.6993(18) Å, β = 101.702(16)° and space group P21/n. Molecular formula: C16H10I1N3O1, unit cell parameters: a = 13.0741(18), b = 4.2988(4), c = 24.974(3) Å, β = 100.040(11)° and space group P21/n. Molecular formula: C18H16N2O1, unit cell parameters: a = 10.8278(7), b = 11.3669(8), c = 13.0687(6)Å, α = 100.716(5)°, β = 106.227(5)°, γ = 104.477(6)° and space group P.†.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H NMR and 13C NMR spectra and crystal data. CCDC 956409–956412. For ESI and crystallographic data in CIF or other electronic format See DOI: 10.1039/c4ra02417f |
| This journal is © The Royal Society of Chemistry 2014 |