Supported Ni–La–Ox for catalytic decomposition of N2O I: component optimization and synergy
Chengliang Li,
Yuesong Shen*,
Shemin Zhu* and
Shubao Shen
College of Materials Science and Engineering, State Key Laboratory of Materials-Oriented Chemical Engineering, Nanjing Tech University, No. 5, Xinmofan Road, Nanjing 210009, P.R. China. E-mail: sys-njut@163.com; Tel: +86-25-83587927
First published on 17th June 2014
Abstract
A series of Ni–La–Ox complex oxides with Ni/La integer molar ratios from 1 to 10 supported on pretreated cordierite ceramics were prepared by an impregnation method and tested for direct catalytic decomposition of N2O. The Ni–La–Ox complex oxides showed considerable synergy in N2O decomposition compared with pure nickel oxide, and the supported Ni–La–Ox complex oxides exhibited much better catalytic performance in reactions. The promotional effects of material structure and synergy of Ni- and La-based oxides on catalytic performance for N2O decomposition were systematically studied through characterization by XRD, N2-BET, H2-TPR, N2O-TPD and XPS. The results show that the NiO and LaNiO3 are major active solid-phases for catalytic decomposition of N2O; the synergetic action between NiO and LaNiO3 promotes oxygen mobility and desorption, and stabilizes the active site of NiII. Furthermore, a new reaction mechanism for N2O decomposition over the supported Ni–La–Ox catalysts is proposed.
1. Introduction
Nitrous oxide (N2O) is a long-lived trace gas, its atmospheric residence time is up to 100–200 years, and its annual growth rate is 0.2–0.3%.1–4 N2O exhibits a harmful global warming potential (GWP) 310 times higher than CO2.4–6 The continuous increase of atmospheric N2O concentration not only caused global warming but also destroyed ozone layer with a subsequent increase in the amount of solar UV-B radiation reaching the earth.6,7 Therefore, how to effectively control and remove N2O has become a major environmental issue of worldwide common concern.Major N2O emission sources are including agricultural soil mining, industrial production of adipic acid, nitric acid and fertilizer, chemical processes using nitric acid as oxidant, coal combustion in fluidized bed, vehicle emissions, and byproduct formation in NH3-SCR of NOx etc., correspondingly current treatment methods of N2O are mainly including thermal decomposition, direct catalytic decomposition, SCR, and as a raw material for producing phenol and nitric acid.4,8,9 Among them, the direct catalytic decomposition method, with merits of relatively low reaction temperature, no reductant consumption, low operating costs, and similar air component decomposition products of N2 and O2, has been regarded as the most promising technology for N2O removal.4,10
As the technological core of the catalytic decomposition method, catalyst has been attracting wide attention. Various catalysts such as noble metals including Ru and Rh etc.,11–15 metal oxides including Co3O4, CuO and Fe3O4 etc.,16–21 supported metal oxides including Fe2O3/Al2O3, CoO/Al2O3 and NiO/mullite etc.,22–26 ion-exchanged zeolites including ZSM-5, ZSM-11 and USY etc.,27–32 and hydrotalcites33–35 are being evaluated for catalytic decomposition of N2O. Although the noble metal catalysts exhibit excellent catalytic performance at low temperature, only a few of them are active and stable enough for N2O decomposition under industrial conditions, because their catalytic activities are seriously inhibited by O2, NO and H2O. Moreover, oxygen atoms formed by N2O decomposition may cause catalyst deactivation.36 In addition, the noble metals are very expensive and their thermal stabilities are poor in the higher temperatures.37 Anyway, the noble metals are not suitable for large-scale industrial applications. The zeolite catalysts exhibit higher decomposition rate of N2O at higher temperatures, while sulfates are easily formed on the surface of the zeolite, consequently resulting in catalytic activity decrease.
Development of catalysts for N2O decomposition aims at the achievements of high catalytic activity, outstanding anti-poisoning ability and long-term cyclic stability at the lower temperature. The reaction mechanism of N2O decomposition with the catalysts is generally considered as a charge donation from catalyst to the anti-bonding orbital of N2O, consequently weakening the N–O bond and decomposing N2O.17 The removal of adsorbed oxygen is the rate-determining step for such a reaction. Hence, the improvement of the oxygen storage capacity and mobility is urgently required in the reaction.38 Catalysts doped with rare-earth oxides often affect the mobility of oxygen on their surfaces. Russo et al.39 studied several LaBO3 (where B is Cr, Mn, Fe, and Co) perovskite-type oxides prepared via solution combustion synthesis, and reported that the LaCoO3 showed the best activity, with 50% N2O conversion at 728 K and 763 K in the absence and presence of 5% of oxygen, respectively. Before this work, we have made a survey of various mixed metal oxide catalysts for N2O decomposition, and found that most researches were concentrated on cobalt-based or copper-based catalysts because of their excellent catalytic activities.18,19,21,40–43 Xue et al.17 reported that the addition of CeO2 into the Co3O4 catalyst could promote catalytic activity, and the CoCe0.05 exhibited full N2O conversion at 563 K. Asano et al.18 reported that a potassium-doped Co3O4 catalyst, with the K/Co molar ratio of 0.02, obtained full conversion of N2O at 573 K. Zhou et al.21 reported that a Cu0.67Ce0.33Oy catalyst exhibited the full N2O conversion at 698 K. As mentioned above, a large number of investigations on catalyst for N2O decomposition were concentrated on pure active components, particularly in powder scale. Anyway, development of a catalyst that is able to apply in real gas streams is one of the great challenges in N2O emission control. As studied by Kapteijn et al.,8 supported metal oxides were not as frequently studied as pure mixed oxides, but for industrial applications the supported metal oxides might be better suited due to their higher dispersion and low cost. However, the catalytic activities of the metal oxides would significantly decrease when they were loaded on carriers for industrial applications. For instance, the full N2O conversions of the Fe2O3/Al2O3 and CoO/Al2O3 reached at 1023 K and 833 K,25,26 respectively. Thereby how to improve the catalytic activity of the supported metal oxides at lower temperatures has become the most important scientific problem.
Besides Co- and Cu-based oxide catalysts, nickel-based oxide catalysts also exhibit good catalytic performance for N2O decomposition, especially for the supported Ni-based oxide catalyst. Lan et al.44 reported that a supported catalyst of NiO/mullite showed full N2O conversion at 753 K. Wang et al.45 studied a catalyst of Ni–Co–Ox supported on the modified cordierite honeycomb ceramic support, the results also indicated that the catalyst showed good catalytic activity and stability in N2O decomposition. As stated above, the supported Ni-based oxide catalysts are promising for N2O decomposition at lower temperature. In this work, a novel supported catalyst of Ni–La–Ox complex oxide, with a pretreated cordierite honeycomb ceramic as carrier, was systematically studied for N2O decomposition in order to obtain the full N2O conversion at much lower temperature. Moreover, the promotional effects of material structure and synergy of Ni- and La-based oxides on catalytic performance for N2O decomposition were mainly studied.
2. Experimental
2.1 Catalyst preparation
A series of Ni–La–Ox complex oxides with 1–10 Ni/La integer molar ratio was prepared by thermal decomposition of Ni–La nitrate solution, which was made of Ni(NO3)2 (Xilong Chemical Co., Ltd, AR 98%) and La(NO3)3 (Sinopharm Chemical Reagent Co., Ltd, AR 99.8) under vigorous stirring at room temperature for 3 h. Then the Ni–La nitrate solution was dried at 333 K for 12 h in air, followed by calcination at 823 K for 2 h. The obtained Ni–La–Ox complex oxides were thus referred to as NixLa (x stands for the molar ratio of Ni/La).2.2 Preparation of supported catalysts
In order to simulate real monolith catalysts for industrial applications and reduce the amount of metal oxide active component, some pretreated cordierite honeycomb ceramics without catalytic ability (as shown in Fig. 2) were crushed into the particle sizes of 6–10 mesh to load uniformly dispersed Ni–La–Ox complex oxides by impregnation method. Ceramic pretreatment via acid pickling technology was washing with boiling 10 wt% HNO3 solutions, and acid pickling temperature was 373 K while the duration time was 30 min. Then the ceramics were washed by distilled water until the pH was neutral, followed by dried at 353 K for 24 h. Subsequently the treated ceramics were immersed into the Ni–La nitrate solution for 1 h, then the supported catalysts were obtained about 10% mass loading amount of Ni–La–Ox complex oxides by drying at 333 K for 3 h, followed by calcination at 823 K for 2 h. The supported catalysts are referred to as S–NixLa (x indicates the Ni/La molar ratio).2.3 Characterization techniques
The X-ray diffraction (XRD) patterns were recorded in the 2-theta from 10° to 80° (0.6° min−1) using X-ray diffractometer (Rigaku DMAX-RB) with a radiation of Cu Kα (λ = 1.5406 Å). The crystal phases were confirmed according to the JCPDS reference. The unit cell parameters were calculated by refining the peak positions of the XRD patterns with a least squares refinement method using the CELREF program.46 The JADE 6.5 program was used to determine peak positions.The environmental scanning electron microscopy (ESEM) experiment was carried out on a JEOL (JSM-5900, Japan) instrument using 10 kV acceleration voltages to determine the morphology and particle size of the Ni4La sample.
The textural properties of the NixLa samples were evaluated from the liquid N2 adsorption–desorption isotherms obtained at 77 K over the whole range of relative pressures, using a Micromeritics ASAP 2020 automatic equipment on samples previously outgassed at 473 K for 2 h. The Brunauer–Emmett–Teller (BET) method was performed to estimate the specific surface area of the NixLa samples. Total pore volumes were determined using the t-plot method, and average pore diameter distributions were derived using the BJH method.
The hydrogen temperature-programmed reduction (H2-TPR) profiles were obtained on a semiautomatic Micromeritics TPD/TPR 2900 apparatus. The samples were pre-treated, before the reduction measurement, at 423 K for 0.5 h in a helium flow, and then cooled to 323 K. Reduction profiles were obtained by passing a 10% H2/Ar flow at a flow rate of 50 mL min−1 through the sample around 30 mg. The temperature was increased from 323 K to 1173 K at a rate of 5 K min−1 and the amount of hydrogen consumed was determined as a function of the temperature.
The selected NiLa, Ni2La, Ni4La, Ni6La, Ni8La and Ni10La samples were analyzed by the temperature-programmed desorption of N2O (N2O-TPD). The samples consisted of solid particles in 25–40 mesh range. Prior to N2O-TPD, the samples around 100 mg were exposed to 30 mL min−1 of helium held at 823 K (10 K min−1) for 1 h, then switched to N2O flow (30 mL min−1) and isothermal treatment at this temperature for 0.5 h, followed by cooling down to 323 K in N2O flow. Afterwards, helium was fed to the reactor at 10 mL min−1 flow and kept flowing for 0.5 h in order to remove any excess oxygen species. The sample was then heated up to 873 K (10 K min−1) under helium flow (10 mL min−1), O2 desorbed during the heating was recorded with the change of temperature.
The X-ray photoelectron spectroscopy (XPS) experiments were carried out on a RBD upgraded PHI-5000C ESCA system (Perkin Elmer) with Mg Kα radiation (hν = 1253.6 eV) or Al Kα radiation (hν = 1486.6 eV). In general, the X-ray anode was run at 250 W and the high voltage was kept at 14.0 kV with a detection angle at 54°. The pass energy was fixed at 23.5, 46.95 or 93.90 eV to ensure sufficient resolution and sensitivity. The base pressure of the analyzer chamber was about 5 × 10−8 Pa. The sample was directly pressed to a self-supported disk (10 × 10 mm) and mounted on a sample holder then transferred into the analyzer chamber. The binding energies of Ni 2p, La 3d and O 1s core levels were determined, referencing to the binding energy of adventitious C 1s signal at a binding energy of 284.6 eV, which gives an accuracy of ±0.1 eV. The data analysis was carried out by using the RBD AugerScan 3.21 software provided by RBD Enterprises and XPSPeak4.1 provided by Raymund W. M. Kwok (The Chinese University of Hong Kong, China). A standard Shirley background and Gaussian (Y%)–Lorentzian (X%) were used for each component.
2.4 Activity measurement and intrinsic kinetic evaluation
The activity measurements were carried out in a fixed-bed reactor using 5 mL catalysts as shown in Fig. 1. The inner diameter of the quartz tube reactor was 10 mm. The S–NixLa samples were placed into the quartz reactor for catalytic decomposition of 5000 ppmv N2O in reaction temperature range of 523–723 K (50 K per each testing temperature) at GHSV of 2400 h−1. The concentrations of N2O at the outlet of reactor were analyzed online by a gas chromatography (Shimadzu GC 2014) equipped with thermal conductivity detector (TCD) and Porapac Q column after 20 min reaction at each temperature. | ||
| Fig. 1 The diagram of catalytic activity test system. | ||
The catalytic activity of S–NixLa samples were evaluated in terms of N2O conversion (XN2O) according to following equation:
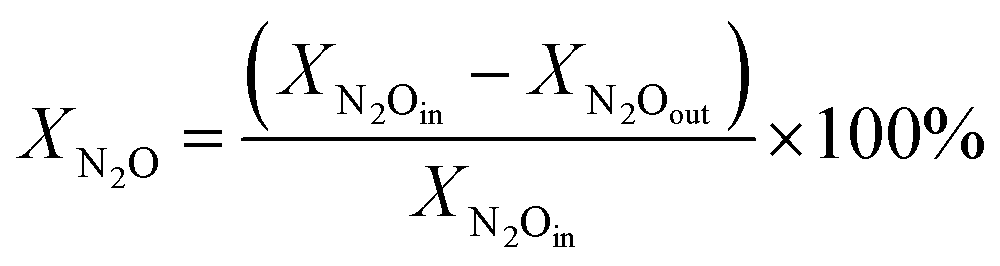 | (1) |
The decomposition of N2O was confirmed to be the first-order in N2O and zero-order in O2 concentration.47 The first-order rate (k), the apparent activation energy (Eapp), and the turnover frequency (TOF) were calculated according to the following equations reported in ref. 48 and 49:
 | (2) |
 | (3) |
 | (4) |
The specific reaction rate (rs, mol s−1 g−1 bar−1) and the intrinsic reaction rate (ri, mol s−1 m−2 bar−1) were calculated according to as reported in ref. 50. FN2O is the N2O molar flow rate at the inlet of the reactor.
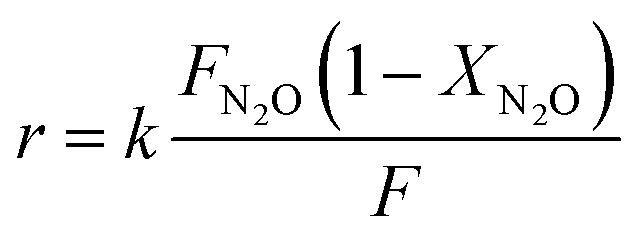 | (5) |
Intrinsic kinetic was estimated using only data corresponding to N2O conversion between 10% and 30%. Furthermore, the Koros–Nowak test51 was performed over S–Ni4La sample with the result verifying that the reaction conditions applied in the present work were located in the kinetic regime. The Koros–Nowak criterion for this reaction according to the following equations:
| ρ = F/mcatfm | (6) |
3. Results and discussion
3.1 Catalytic activity results
Fig. 2 shows the profile of N2O conversion versus temperature over S–NixLa samples. The pure NiO supported catalyst with the same mass loading amount and particle size as those of the S–NixLa catalysts was tested for catalytic decomposition of N2O in order to deeply reveal synergy of the Ni- and La-based oxide components in reaction. The activity of the pure NiO supported catalyst was low, and its N2O conversion at 723 K was less than 30%. The doping of La generated more oxygen vacancies than bulk NiO catalyst on the surface of sample, and has a promotional effect on the catalytic activity. Thus, the S–NixLa catalysts exhibit better catalytic activity than bulk NiO supported catalyst. Compared with the catalytic performance of the pure NiO supported catalyst, the treated cordierite honeycomb ceramics exhibited almost no activity in reaction. It illustrates that herein the cordierite ceramics only play the role of loading and dispersing active NixLa while have no effect on catalytic decomposition of N2O. So the effect of the treated cordierite ceramics on catalytic performance will be ignored in the following analysis and discussion. | ||
| Fig. 2 Conversion of N2O over supported NixLa catalysts with different Ni/La ratios. Conditions: 5000 ppm N2O, balance N2, GHSV = 2400 h−1. | ||
The S–NixLa catalysts showed slight loss of activity in the temperature of 523–573 K. This loss may be related to physical adsorption of N2O before 573 K in the heating process while no such phenomenon occurred in cooling process. This idea is supported by the Weber et al.'s study.52 The total activity increased with increasing reaction temperature, particularly in temperature range from 573 K to 723 K. However, the actual catalytic activities of the S–NixLa catalysts were different to each other with increasing La content in the series. The N2O was decomposed completely at the lowest temperature of 708 K, which was obtained by the S–Ni4La catalyst. The catalytic activity of S–Ni4La and S–Ni8La samples were 88.4% and 83.2% at the temperature of 673 K, respectively. However, the catalytic activities were almost same for S–Ni4La and S–Ni8La samples at 698 K. This may be due to the crystallite size of NiO in S–Ni8La sample is smaller than S–Ni4La sample. N2O completely decomposition for S–Ni8La catalyst obtained at 713 K, which is higher than S–Ni4La sample. This indicated that the crystallite size is not the determining factor for catalytic activity.
In the heterogeneous catalytic reaction, it is well known that the mass transfer limitations play an important role on the reaction rate.53 The heat- and mass-transfer limitations may be appeared during the reaction kinetic test. For this reason, the Koros–Nowak test was carried on 10 wt% and 12 wt% S–Ni4La samples, respectively (Table 1). Fig. 3 shows the N2O conversion over 10 wt% and 12 wt% S–Ni4La samples. It is clearly observed that the reaction obeyed the Koros–Nowak criterion, which demonstrated that our present work are not affected by the heat- or mass-transfer limitations and located in the kinetic regime.
| Samples | Metal content (wt%) | Catalyst amount (g) |
|---|---|---|
| S–Ni4La-10% | 10.4 | 3.65 |
| S–Ni4La-12% | 12.6 | 3.01 |
 | ||
| Fig. 3 Koros–Nowak tests for S–Ni4La-10% and S–Ni4La-12% samples. | ||
Considering that the real metal oxides loadings for the S–NixLa samples were different, the intrinsic kinetic parameters (rate constant, reaction rate and turnover frequency) were compared as interpreted in Fig. 4. The intrinsic reaction rates and rate constants of N2O decomposition are depicted as a function of 1/T (Arrhenius plots, Fig. 4a and b). It can be seen that per m2 of Ni4La surface area was able to convert the largest number of N2O at per unit time, indicating that the S–Ni4La possesses the highest N2O conversion rate. Moreover, the effect of the Ni/La molar ratio on the reaction rate was not promoted as the Ni/La molar ratio increased. It implies that the interaction between Ni- and La-based oxide components plays key role in reaction; more or less La additives in S–NixLa may be not helpful for N2O decomposition.
 | ||
Fig. 4 (a) Arrhenius plots ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ri versus 1/T over the S–NixLa samples; (b) Arrhenius plots lnk versus 1/T over the S–NixLa samples; (c) turnover frequencies (TOF) of the N2O decomposition over the S–NixLa samples. N2O direct decomposition versus reaction temperature corresponding to the N2O conversion between 10% and 30%. ri versus 1/T over the S–NixLa samples; (b) Arrhenius plots lnk versus 1/T over the S–NixLa samples; (c) turnover frequencies (TOF) of the N2O decomposition over the S–NixLa samples. N2O direct decomposition versus reaction temperature corresponding to the N2O conversion between 10% and 30%. | ||
The turnover frequency (TOF) values for the S–NixLa catalysts are presented in Fig. 4c as a function of T. The results clearly demonstrate that per molar cationic ions of S–Ni4La catalyst was able to convert the largest molar number of N2O at per unit time, further indicating that the S–Ni4La possesses the best catalytic performance.
For better comparison of their catalytic performances, the intrinsic kinetic parameters have been tentatively calculated at 610 K. The calculation results of the kinetic parameters are listed in Table 2. More obvious comparisons can be obtained from the reaction rate values expressed per m2 (per g) which led to the highest value on S–Ni4La catalyst. In addition, the TOF shows that the S–Ni4La catalyst had the highest values. By the comparison, the reaction rates of S–NixLa samples were higher than those values reported in the literature. This may be due to that the mobility of oxygen species on the NixLa surface is more easily.
| Catalyst | k/× 10−5 mol s−1 g−1 bar−1 | Specific reaction rate/× 10−7 mol s−1 g−1 bar−1 | Intrinsic reaction rate/× 10−8 mol s−1 m−2 bar−1 | TOF/× 10−5 s−1 | Eapp/kJ mol−1 | Specific surface area/m2 g−1 |
|---|---|---|---|---|---|---|
| a Measured at 808 K.b Measured at 848 K. | ||||||
| NiLa | 9.74 | 3.99 | 2.69 | 5.87 | 72.9 | 14.8 |
| Ni2La | 6.89 | 2.96 | 2.01 | 3.74 | 124.8 | 14.8 |
| Ni3La | 12.3 | 4.73 | 3.02 | 6.02 | 44.8 | 15.7 |
| Ni4La | 16.6 | 5.81 | 3.71 | 7.74 | 41.8 | 15.6 |
| Ni5La | 6.54 | 4.01 | 2.63 | 4.51 | 112.8 | 15.2 |
| Ni6La | 7.52 | 3.21 | 1.35 | 3.32 | 115.7 | 23.7 |
| Ni7La | 5.33 | 2.37 | 0.88 | 2.22 | 110.3 | 27.2 |
| Ni8La | 8.37 | 3.51 | 1.28 | 3.57 | 145.2 | 27.4 |
| Ni9La | 7.07 | 3.04 | 1.69 | 2.98 | 124.4 | 17.9 |
| Ni10La | 14.4 | 5.31 | 2.14 | 6.02 | 69.2 | 24.9 |
| La0.8CoO3 (ref. 50) | 0.20a | 0.84a | 177b | |||
The apparent activation energies on S–NixLa samples were all less than 150 kJ mol−1, which were smaller than 177 kJ mol−1 reported in the literature. These values are lower than the energy requires to break the N–O bond in the N2O molecule without catalyst participation (250–270 kJ mol−1), which requires about 898 K.54 Therefore, through above intrinsic kinetic evaluations, it can be found that the S–Ni4La reveals a maximum synergetic effect between Ni- and La-based oxide components in reaction.
3.2 Textural characteristics
Table 2 lists The BET specific surface areas of the NixLa samples. The BET values range from 14.8 m2 g−1 to 27.4 m2 g−1. The S–Ni8La catalyst does not exhibit the optimal catalytic activity, even though it has the biggest specific surface area 27.4 m2 g−1. According to comparative analysis of specific surface area and catalytic activity, there is no direct relationship between their change trends. On the other words, the specific surface area is not the sole determining parameter here.16,18 Meanwhile, there is also no direct relationship between catalytic activity and total pore volume as shown in Fig. 5. However, it can be found that the Ni4La possesses the smallest average pore diameter about 17.4 nm, which may be the most appropriate size for mass transfer channel in reaction.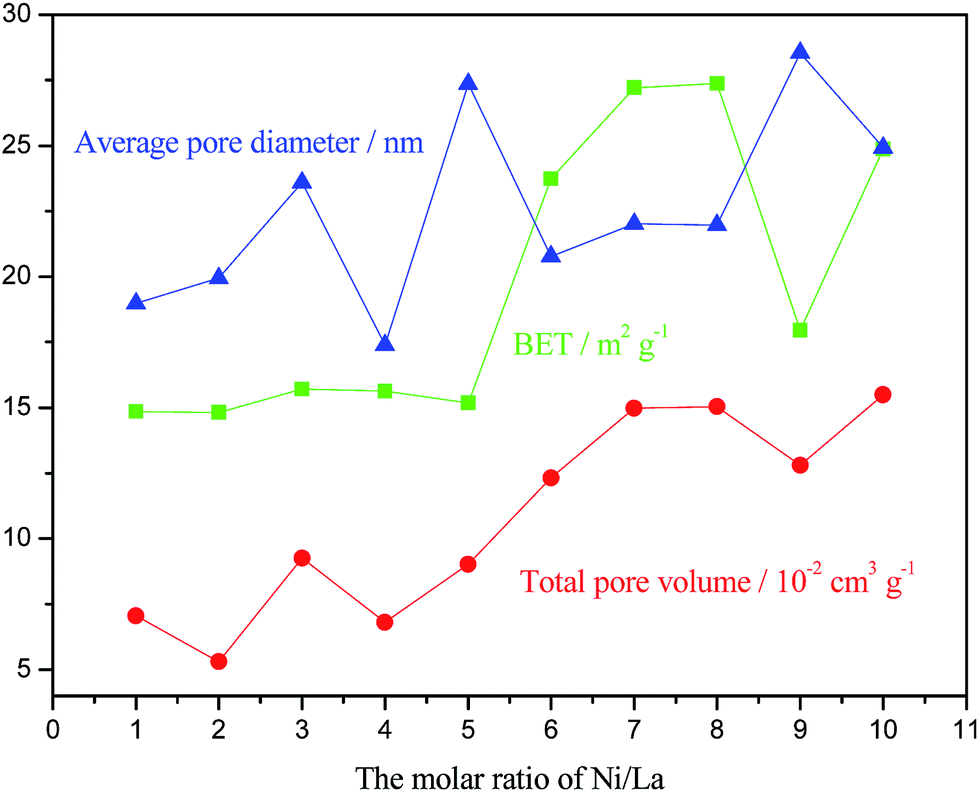 | ||
| Fig. 5 Textural properties of NixLa samples. | ||
3.3 XRD analysis
Fig. 6 shows the X-ray diffractograms of the NixLa and pure NiO powders calcined at 823 K before reaction. All the reflections of the NixLa samples provide typical diffraction patterns for the cubic NiO (PDF 47-1049, marked as ▼) and perovskite-type LaNiO3 (PDF 34-1181, marked as ●). Decreased intensity was observed in the reflections from cubic NiO with increased La concentration in the samples. Compared to the diffraction peaks of pure NiO sample, the NiO diffraction peaks of the NixLa samples were wider, indicating that the La additives minimize the crystal size of NiO. The formation of LaNiO3 structure means that the part of NiII was oxidized into NiIII, and the size of the NiIII cation (0.62 Å) is smaller than that of the LaIII cation (1.06 Å) inside the La2O3 structure, implying that the presence of LaNiO3 perovskite structure could significantly promote the mobility of oxygen on the catalysts surface. In addition, typical diffraction patterns for the La2O3 (PDF 73-2141, marked as ■) and La(OH)3 (PDF 36-1481, marked as ^) were able to be found when the Ni/La molar ratio was less than 4. The presence of La(OH)3 may be due to the reaction between La2O3 and H2O as follows: La2O3 + H2O → La(OH)3, because rare-earth oxides are easily hygroscopic when they were exposed to air conditions.55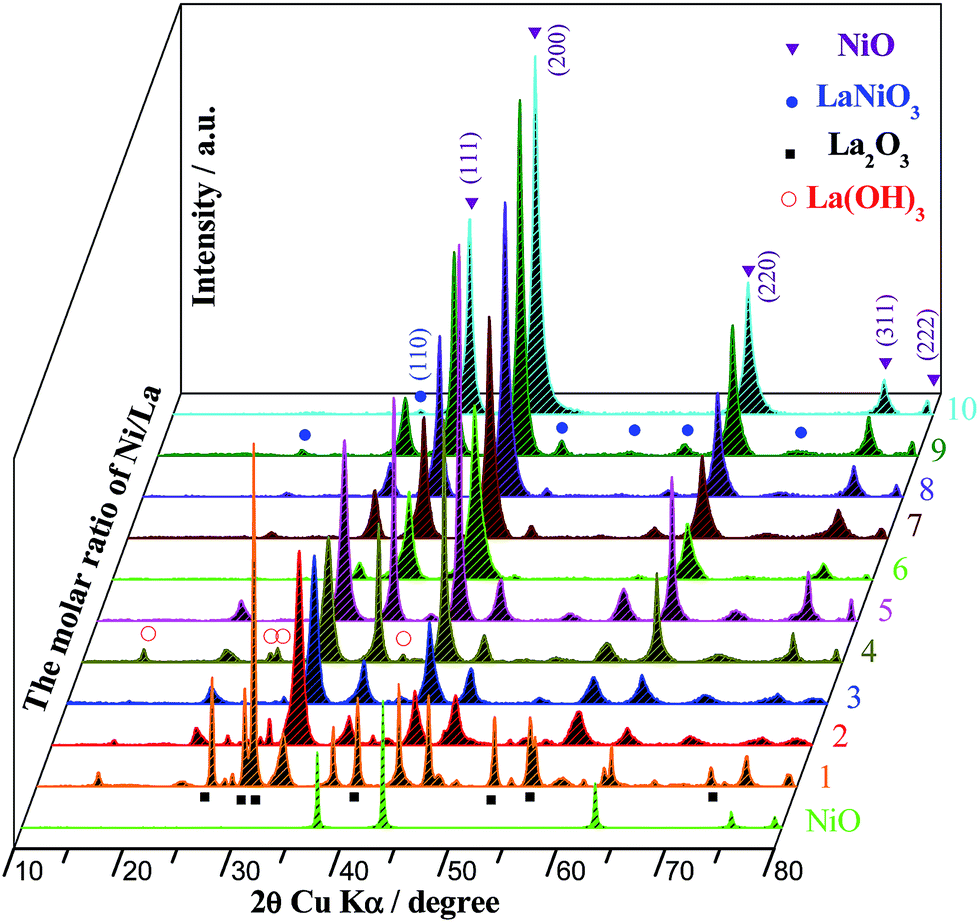 | ||
| Fig. 6 XRD patterns of the NixLa and pure NiO samples. | ||
To facilitate comparisons, the percentage compositions of each crystal phase in the NixLa samples were obtained by Relative Intensity Ratio (RIR) method using JADE 6.5 program as shown in Fig. 7. In terms of the La2O3 crystalline phase, the diffraction peaks were also become more intensive with the increasing proportion of La. The peaks of La2O3 are quite weak in Ni4La sample and totally disappear in Ni5La sample. This demonstrated that quite low amount of La2O3 existed in Ni4La sample, which corresponded to the calculated result about 0.5%. When x = 1, the percentage composition of La2O3 is the highest about 63.8%.
 | ||
| Fig. 7 The percentage compositions of each phase in the NixLa samples. | ||
The grain size of NiO in the Ni4La sample is small (about 15 nm), and the presence of appropriately amount of LaNiO3 structure increased the mobility of O species. Thus, the supported Ni4La catalyst show the best catalytic performance. Compared to the solid-phase structure and the catalytic activity of the pure NiO sample, the LaNiO3 should be a major active phase for catalytic decomposition of N2O, especially for the samples of Ni5La, Ni6La, Ni7La, Ni8La, Ni9La and Ni10La. To the best of our knowledge, the NiO is also a major active component for catalytic decomposition of N2O, additionally the catalytic activities of the catalysts were not increased with the increased percentage composition of LaNiO3 or NiO, and so the synergy of NiO and LaNiO3 gives rise to catalytic decomposition of N2O.
To determine whether or not solid solutions were formed in mixed NixLa samples, unit cell parameters were calculated from Bragg's angles, and results are shown in Table 3. For NiLa sample, the unit cell parameters of the LaNiO3 are bigger than others. This may be due to the presence of La2O3 promoting the growth of crystalline grain, suggesting that the substitution of La resulted in the distortion of lattice, especially for the Ni4La sample.
| NiLa | Ni2La | Ni3La | Ni4La | Ni5La | ||
|---|---|---|---|---|---|---|
| NiO | a/Å | 4.1784 ± 0.0014 | 4.1821 ± 0.0130 | 4.1776 ± 0.0011 | 4.1791 ± 0.0007 | 4.1776 ± 0.0003 |
| LaNiO3 | a/Å | 5.5003 ± 0.0099 | 5.4418 ± 0.0128 | 5.4604 ± 0.0045 | 5.4451 ± 0.0122 | 5.4533 ± 0.0111 |
| c/Å | 6.5940 ± 0.0014 | 6.5740 ± 0.0050 | 6.5926 ± 0.0008 | 6.5646 ± 0.0038 | 6.5704 ± 0.0135 |
| Ni6La | Ni7La | Ni8La | Ni9La | Ni10La | ||
|---|---|---|---|---|---|---|
| NiO | a/Å | 4.1760 ± 0.0015 | 4.1794 ± 0.0004 | 4.1779 ± 0.0011 | 4.1789 ± 0.0011 | 4.1777 ± 0.0010 |
| LaNiO3 | a/Å | 5.4739 ± 0.0074 | 5.4562 ± 0.0102 | 5.4527 ± 0.0040 | 5.4300 ± 0.0249 | 5.4525 ± 0.0109 |
| c/Å | 6.5243 ± 0.0402 | 6.5599 ± 0.0080 | 6.5593 ± 0.0016 | 6.5857 ± 0.0052 | 6.5718 ± 0.0053 |
It can be observed that the diffraction peaks of NiO phase shift toward higher angles when compared with the standard JCPDS card as shown in Fig. 8. This may be due to the substitution of La, leading to the distortion of lattice. On the contrary, the diffraction peaks of LaNiO3 phase shift toward lower diffraction angles. As mentioned above, perovskite-type LaNiO3 formed in which LaIII is substituted by NiIII. Additionally, Wolska et al.56 reported that the charge neutrality could be achieved as substitution of O2− by OH−, also could cause the distortion of lattice and leading the shift of diffraction angles.
 | ||
| Fig. 8 The diffraction angles of NiO (200) and LaNiO3 (110). | ||
3.4 Micro-morphology
Fig. 9a and b show the microstructure morphology of the Ni4La sample. The Ni4La microstructure looks very loose, such as coral-like structure. It can be seen by ESEM that the equivalent diameter of nano-particle is about 20–25 nm. There are a large number of irregular interstitial holes existed in staggered accumulation of nano-particles. Fig. 9c and d show the surface morphology of bare ceramic honeycomb support. It can be seen that the images of bare ceramic honeycomb presents sheet-like structure. In addition, it is observed that a lot of micro pores are found on the surface of bare ceramic honeycomb. This may be have a great contribution to the activities of Ni–La–Ox supported catalysts. The photographs of supported Ni4La catalyst are shown in Fig. 9e and f. It can be clearly seen that the presence of Ni–La–O mixed metal oxides, and dispersed homogeneously on the surface of the support. | ||
| Fig. 9 SEM photograph of Ni4La mixed metal oxides (a and b); SEM photograph of ceramic honeycomb support (c and d); SEM photograph of supported Ni4La catalyst (e and f). | ||
3.5 H2-TPR
Asano et al.18 reported that the reducibility enhancement of mixed metal oxides can effectively promote the catalytic activity of N2O decomposition. The H2-TPR measurements were performed over the NiLa, Ni2La, Ni4La, Ni6La, Ni8La and Ni10La samples in order to determine the reducibility and its correlation with catalytic activity. The H2-TPR profiles of the samples are shown in Fig. 10. The weak peaks appearing at low temperature around 373–573 K are named α peak, which were attributed to the reduction of surface absorbed O species and dispersed NiO. As mentioned above, the ionic radius of NiII is smaller than LaIII, and NiII incorporated into the lattice of La2O3 to form LaNiO3 perovskite structure. The formation of LaNiO3 leads to generation of oxygen vacancies, which adsorb oxygen species easily. Therefore, very active oxygen species are formed, which are reduced by H2 at low temperature. | ||
| Fig. 10 H2-TPR profiles of the NixLa samples with different Ni/La molar ratio. Conditions: 10% H2/Ar, 50 mL min−1 and ramping rate of 5 K min−1. | ||
The β peak appearing at around 673 K enhances as Nickel content increases, which corresponds to the reduction of NiO to metallic nickel. The reduction peak of pure NiO sample appeared at around 690 K (data was not shown). This indicated that the reduction ability of NixLa samples were better than bulk NiO sample. Thus, increased the ability of catalysts to accept electronic, and the NixLa catalysts show better catalytic activity. According to the research of Zhou et al.,38 the reduction process of NiO usually follows below: NiO → Niδ+ → Ni0, because the crystalline size of pure NiO is big. However, the stepwise reduction of NiO to metallic nickel is not observed in NixLa samples. This illustrates that La additives minimize the crystallite size of NiO. As analyzed above, it can be further implied that the interaction between NiO and LaNiO3 promotes the oxygen mobility and leads to only one main peak observed.
In general, the reduction process of LaNiO3 perovskite oxide follows two consecutive steps expressed by eqn (7) and (8).57 In the first step about 693 K, NiIII ions stabilized in the LaNiO3 structure are reduced into NiII, and form an oxygen deficient LaNiO2.5 structure. In the second step about 823 K, the LaNiO2.5 is reduced to metallic nickel and La2O3. For the Ni2La and Ni4La samples, the shoulder peak is observed and named β1, indicating that the first reduction peak of LaNiO3 was overlapped with the reduction peak of NiO. With further increase of x values, the β1 peak disappears and the reduction of NiO species in the NixLa samples presents only one main peak. Furthermore, the H2 consumption of γ peak attributed to the second reduction step of LaNiO3 decreased with the increase of x values from 2 to 10, which are in accordance with the content of LaNiO3 in NixLa samples calculated by XRD patterns.
| LaNiO3 + 1/2H2 → LaNiO2.5 + 1/2H2O | (7) |
(Low-temperature peak)
| LaNiO2.5 + H2 → 1/2La2O3 + Ni + H2O | (8) |
(High-temperature peak)
For NiLa sample, the β1 peak was not observed. This indicated that only few amount of LaNiO3 exist in the NiLa sample. However, the area of γ peak is larger than others. The appearing of γ peak in NiLa sample must be attributed to the reduction process of other species. According to the study of G. Wrobel et al.,58 when NiO dispersed on the surface of solid solution, strong interaction between free NiO and solid solution is achieved. In the case of NiLa sample, there are large amount of La2O3 contained in the sample, there must be of some NiO dispersed on the surface of La2O3, so their interaction product may be reduced at higher temperature corresponded to the γ peak.
The peak positions change obviously with the increase of x from 1 to 10. This indicated that a proper amount of LaNiO3 can give rise to significant changes of catalyst structure and thereby enhance them reduction ability. For Ni4La sample, the onset temperatures of the peaks shift to high values. This may be due to the interaction between NiO and LaNiO3, further indicating that the formation of LaNiO3 solid solution is effective in stabilizing the lower oxidation state of NiII. Thus, it is easy that the electron transfer from the catalyst to N2O molecule.
3.6 N2O-TPD
Catalytic decomposition of N2O usually is considered as an oxidation–reduction reaction mechanism.59 This reaction process based on electron transfer follows a three-step mechanism expressed by eqn (9)–(11). Oxides with some local charge donation properties isolated transition metal ions, and leading to more than one valence act as the active site on the surface.8 In the case of the NixLa samples, NiII serves as active site in the reaction process. It is reported that the removal of adsorbed oxygen is the rate-determining step of N2O decomposition. Therefore, N2O temperature-programmed desorption (N2O-TPD) experiment was performed for the investigation of the relationship between N2O catalytic activity and O2 desorption.| N2O + e−sur → N2 + O−ads | (9) |
| O−ads + N2O → N2 + O−2ads | (10) |
| 2O−ads ↔ O2 + 2e−sur | (11) |
The decomposition of N2O over the catalysts left adsorbed O species on the surface during the cooling down step. Fig. 11 shows the N2O-TPD profiles of the NixLa samples. Several peaks can be seen in the profiles at temperatures from 323 to 873 K. The peak appearing from 373 to 523 K is named α peak, they can be corresponded to desorption of chemically adsorbed oxygen species (O−, O2−) on the oxygen vacancies, which formed during reaction process. Obviously that desorption peaks shift to higher temperature with Ni content increasing in the NixLa mixed oxide, and The α peak of Ni4La sample shifts to low temperature, indicating that the mobility and desorption of O2 are facilitated on the surface of the Ni4La sample, thus the Ni4La sample shows the best catalytic activity for N2O decomposition. Moreover, as the increasing amount of Ni, most of Ni species do not have strong interaction with La, so that the adsorbed O species can be desorbed in the high temperature.
 | ||
| Fig. 11 N2O-TPD profiles of the NixLa samples with different Ni/La molar ratio. | ||
The β peak appeared at around 753 K can be attributed to desorption of subsurface oxygen. In the case of Ni2La, the temperature of β peak shifts to low value, implied that interaction between O species and metal ions results in desorption of subsurface oxygen easily. According to change the trend of N2O-TPD profiles, it can be supposed that another γ peak may appear above 873 K. The attribution of the γ desorption peak is the lattice oxygen, namely, the oxygen that is released by the reduction of NiIII according to the following reaction:60
| 2NiIII + O2− → 2NiII + VO + 1/2O2 | (12) |
For NiLa sample, the peak centered at 723 K is named δ peak. Nevertheless, the δ peak was not appearing at other samples. Combined with the results of XRD analysis, the presence of La2O3 hindered desorption of oxygen, which in agreement with the result of catalytic activity. As mentioned above, the NiII acts as the active site for N2O decomposition. The strength of oxygen bond with active site will directly influence the oxygen mobility. As a result, the desorption of oxygen species can only be achieved at relatively higher temperatures.17
3.7 XPS
XPS measurements were carried out to examine the surface electronic state of the Ni4La sample. Fig. 12 and 13 presented the spectra of Ni 2p, La 3d and O 1s core-level, respectively. The binding energies of Ni 2p1/2, 2p3/2, La 3d3/2, 3d5/2 and O 1s determined by XPS are summarized in Table 4. The Ni/La atomic ratio was determined by the element relative sensitivity factor (RSF) method, and calculated using. The calculation result was 3.58 (fresh catalyst) and 4.42 (treated by N2O), which were close to the design Ni/La atomic ratio of 4.
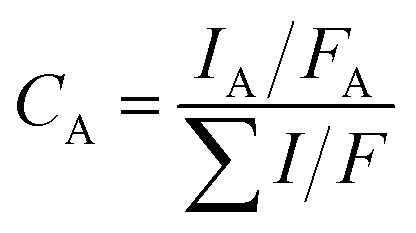 | (13) |
 | ||
| Fig. 12 Ni 2p and La 3d core level spectra of the Ni4La sample at two conditions, (1) fresh catalyst and (2) after exposure of N2O (N2 balance). | ||
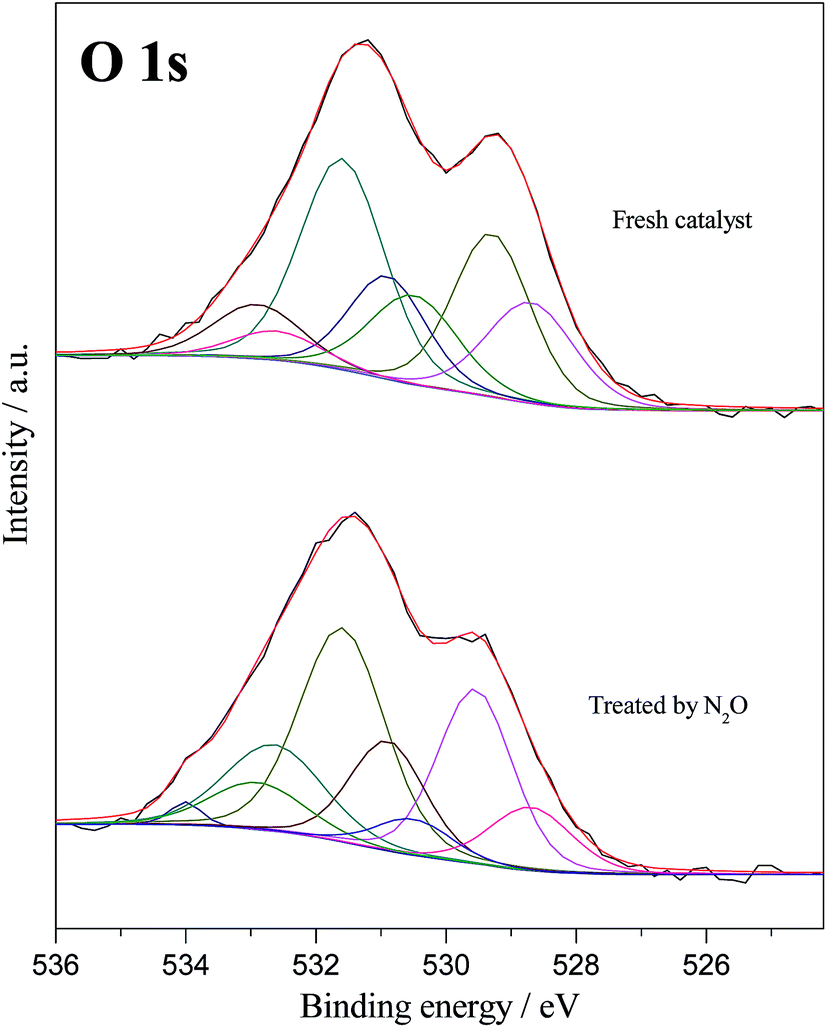 | ||
| Fig. 13 O 1s core level spectra of the Ni4La samples at two conditions: (1) fresh catalyst and (2) after exposure of N2O (N2 balance). | ||
| Peak parameters | Peak identification | |||||
|---|---|---|---|---|---|---|
| Fresh Ni4La catalyst | Treated by N2O | Species | Line | Assignment | ||
| BE (eV) | FWHM | BE (eV) | FWHM | |||
| 871.9 | 4.17 | 871.9 | 4.13 | NiO | 2p1/2 | Core |
| 853.9 | 2.85 | 853.9 | 2.78 | NiO | 2p3/2 | Core |
| 873.1 | 4.04 | 873.2 | 3.98 | Ni2O3 | 2p1/2 | Core |
| 855.8 | 2.47 | 855.8 | 2.51 | Ni2O3 | 2p3/2 | Core |
| 861.4 | 5.43 | 861.4 | 5.22 | Satellite | ||
| 879.1 | 6.12 | 879.1 | 5.04 | Satellite | ||
| 852 | 4.1 | 851.9 | 4.04 | La2O3 | 3d3/2 | Core |
| 855.3 | 4.08 | 855.2 | 3.94 | Shake up | ||
| 835.2 | 3.69 | 835.2 | 3.8 | La2O3 | 3d5/2 | Core |
| 838.5 | 2.6 | 838.4 | 2.6 | Shake up | ||
| 531.6 | 1.52 | 531.6 | 1.82 | NiII–O | 1s | Core |
| 529.3 | 1.4 | 529.5 | 1.36 | La–O | 1s | Core |
| 530.9 | 1.41 | 530.9 | 1.59 | NiIII–O | 1s | Core |
| 532.6 | 1.6 | 532.6 | 2.08 | O ads | 1s | Core |
| 528.7 | 1.6 | 528.7 | 1.49 | O2− | 1s | Core |
| 530.5 | 1.6 | 530.5 | 1.4 | O− | 1s | Core |
| 532.9 | 1.59 | 532.9 | 1.75 | H2O/OH | 1s | Core |
| 534 | 0.58 | Ads-O2 | 1s | Core | ||
Fig. 12 shows the peak fitting results of Ni 2p and La 3d binding energies, which are determined by peak fitting through XPSPeak4.1. As we known that the La 3d3/2 peak overlap Ni 2p3/2 peak,61 and the La 3d core level split into 3d5/2 and 3d3/2 components due to a spin–orbit interaction. Additionally each line split to main line (3d04f0 final state configuration) and a satellite line (3d04f1L final state configuration).62,63 For the fresh and used Ni4La samples, the peaks of Ni 2p appearing at 871.9 eV and 853.9 eV belong to NiII core-level. Moreover, shake up satellite peaks of NiII appear at 861.4 eV and 879.1 eV, respectively. This indicates that the presence of NiO on the catalyst surface. The binding energies of 873.1 eV and 855.8 eV correspond to NiIII 2p1/2 and NiIII 2p3/2 core-level, which related to the formation of LaNiO3 on the surface. The percentage content of NiIII is about 25%. These results demonstrated that only NiO and LaNiO3 species were present on the catalyst surface, which are similar to XRD results.
The spectra were not significantly different among the samples. However, a small change was observed over a variety of peaks that fitted the spectra. This change may be caused by sample preparation during the experiments. Such change of fitting peaks was also observed by Mark C. Biesinger.64 If the samples contain only a small amount of NiII, then the spectrum is very well defined, but if the degree of reduction of NiIII to NiII is high, then the binding energy of Ni 2p3/2 would shift to low values. For the Ni4La sample, the binding energy of Ni 2p3/2 was not shifted after pretreatment by N2O at 723 K. This indicates that the NiII is not oxidized by N2O treatment, further suggesting that the NiII and oxygen vacancies are more stable during the N2O decomposition process. As a result, they are able to enhance desorption and mobility of the oxygen, which is the rate-determining step in such a reaction. This result is in great agreement with the analysis of activity and stability.
The core levels of O 1s spectra for two samples are shown in Fig. 13. The O 1s peaks are complex and this result indicates that the presence of more than one type of oxygen species in the surface. The peak appearing at 529.3 eV can be ascribed to lattice O2− anion in lanthanum oxides.65 According to the literature, the chemical boding in LaNiO3 was not purely ionic, but exhibited covalent (Ni–O and La–O) and metallic (Ni–O–Ni) parts that can explain the presence of two different oxides in the fresh sample.66 The peaks appearing at 530.5 eV and 534 eV in the used sample can be ascribed to adsorbed O− and O2, which were not found in the fresh sample. This indicates that O− and O2 form during the N2O decomposition process, which in agreement with the result of N2O-TPD. In addition, if NiII anion was oxidized to NiIII after pretreatment by N2O, the peak at 531.6 eV would start to vanish. This factor further confirms that the NiII is not oxidized by N2O treatment. As mentioned above, the rare earth oxides are easily hygroscopic when they are exposed to atmospheric conditions.55 The peak appearing at 532.9 eV can be attributed to the adsorbed water species at the surface. Future studies are needed to evaluate the catalyst activity in the presence of O2 and H2O.
4. Discussion
So far, several reaction mechanisms for N2O decomposition have been proposed by different researchers.10,21,59 It is generally accepted that the reaction is an oxidation–reduction process. The surface oxygen vacancies (VO) and metal ions act as the active center, and N2O act as an oxidizing agent in the reducing step. The S–NixLa samples exhibit higher catalytic activity than that of pure NiO supported catalyst. This may be due to the presence of LaNiO3 structure which gives rise to the oxygen non-stoichiometry in the surface. Based on the results obtained, we proposed a possible reactions mechanism as presented in Scheme 1. To the best of our knowledge, N2O molecular is linear structure and it can be adsorbed either through O or N ends.18 As for the case of coordination on N end, the N–O bond order increases and the N–N bond decreases. As a result, NO or NO2 would be the byproducts for N2O decomposition. However, we have not yet detected NO or NO2 species in XPS and GC analysis. Therefore, the coordination takes place on the O end, and product is nitrogen. As is shown in Scheme 1, N2O deposits on the active centers such as oxygen vacancies and NiII with the O atom expressed in eqn (14)–(17). This process involved a transient N2O− specie that formed by the electron from the dxy donor orbital toward 3π* acceptor orbital of the N2O. Subsequently dissociative chemisorption of N2O molecule on the surface active center and release of N2 expressed by eqn (16) and (17). The XPS results also confirm the presence of O2− and O− species.| NiII + N2O → NiII⋯ONN | (14) |
| VO + N2O → VO⋯ONN | (15) |
| NiII⋯ONN → NiIII⋯O− + N2 | (16) |
| VO⋯ONN → VO⋯O2− + N2 | (17) |
 | ||
| Scheme 1 Catalytic mechanism for N2O decomposition over the supported NixLa catalysts. | ||
In terms of the activity result, the presence of LaNiO3 increased the catalytic activity of catalyst. This may be due to the presence of NiIII facilitated the NiIII (NiIII–O−) reduced to NiII, and oxygen removed promptly. However, the activity was not increased with the increased amount of LaNiO3. The removal of adsorbed oxygen is the rate-determining step for N2O decomposition. After the above steps, the formation of O2 in two ways: (i) the two neighboring O− species combined with each other and NiIII was reduced into NiII expressed by eqn (18); (ii) the O2− in vacancy sites interaction with another N2O molecule expressed by eqn (19). These steps involved the electron migration from O− or O2− species to the surface. The results of N2O-TPD and XPS clearly support this hypothesis as discussed.
| 2NiIII⋯O− → 2NiII + O2 | (18) |
| VO⋯O2− + N2O → VO + O2 + N2 | (19) |
In summary, the LaNiO3 enhanced desorption and migration of oxygen, and resulted in high catalytic activity for N2O decomposition over the NixLa supported catalysts.
5. Conclusions
This work has been confirmed that the supported Ni–La–Ox complex oxides are promising catalyst system for N2O decomposition. The S–Ni4La catalyst obtains full N2O conversion at 708 K and displays perfect stability in N2O decomposition. The NiO and LaNiO3 are major active solid-phase structures for catalytic decomposition of N2O, and the promotional mechanism is that the synergetic action between NiO and LaNiO3 promotes oxygen mobility and desorption, and stabilizes the active site of NiII. Future studies are needed to evaluate catalytic performance in the presence of O2 and H2O.Acknowledgements
This work was supported by the National Key Technology Support Program (no. 2012BAE01B03), the National Natural Science Foundation of China (no. 21106071, 51272105), the New Teachers' Fund for Doctor Stations Sponsored by the Ministry of Education of China (no. 20113221120004), the Research Subject of Environmental Protection Department of Jiangsu Province of China (no. 2012016), the Jiangsu Provincial Science and Technology Supporting Program (no. BE2013718, BE2011184), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and Program for Changjiang Scholars and Innovative Research Team in University (no. IRT1146).References
- P. Crutzen and C. Howard, Pure Appl. Geophys., 1978, 116, 497–510 CrossRef CAS.
- M. Kavanaugh, Atmos. Environ., 1987, 21, 463–468 CrossRef CAS.
- G. Centi, S. Perathoner and F. Vazzana, CHEMTECH, 1999, 29, 48–55 CAS.
- J. Perez-Ramirez, F. Kapteijn, K. Schoffel and J. A. Moulijn, Appl. Catal., B, 2003, 44, 117–151 CrossRef CAS.
- H. Rodhe, Science, 1990, 248, 1217–1219 CAS.
- M. A. Wojtowicz, J. R. Pels and J. A. Moulijn, Fuel Process. Technol., 1993, 34, 1–71 CrossRef CAS.
- J. C. Kramlich and W. P. Linak, Prog. Energy Combust. Sci., 1994, 20, 149–202 CrossRef CAS.
- F. Kapteijn, J. RodriguezMirasol and J. A. Moulijn, Appl. Catal., B, 1996, 9, 25–64 CrossRef CAS.
- G. Centi, A. Galli, B. Montanari, S. Perathoner and A. Vaccari, Catal. Today, 1997, 35, 113–120 CrossRef CAS.
- S. Kumar, A. Vinu, J. Subrt, S. Bakardjieva, S. Rayalu, Y. Teraoka and N. Labhsetwar, Catal. Today, 2012, 1998, 125–132 CrossRef PubMed.
- K. Yuzaki, T. Yarimizu, K. Aoyagi, S. Ito and K. Kunimori, Catal. Today, 1998, 45, 129–134 CrossRef CAS.
- J. Haber, M. Nattich and T. Machej, Appl. Catal., B, 2008, 77, 278–283 CrossRef CAS PubMed.
- V. G. Komvokis, G. E. Marnellos, I. A. Vasalos and K. S. Triantafyllidis, Appl. Catal., B, 2009, 89, 627–634 CrossRef CAS PubMed.
- H. Beyer, J. Emmerich, K. Chatziapostolou and K. Koehler, Appl. Catal., A, 2011, 391, 411–416 CrossRef CAS PubMed.
- S. S. Kim, S. J. Lee and S. C. Hong, Chem. Eng. J., 2011, 169, 173–179 CrossRef CAS PubMed.
- F. J. Perez-Alonso, I. Melian-Cabrera, M. L. Granados, F. Kapteijn and J. L. G. Fierro, J. Catal., 2006, 239, 340–346 CrossRef CAS PubMed.
- L. Xue, C. Zhang, H. He and Y. Teraoka, Appl. Catal., B, 2007, 75, 167–174 CrossRef CAS PubMed.
- K. Asano, C. Ohnishi, S. Iwamoto, Y. Shioya and M. Inoue, Appl. Catal., B, 2008, 78, 242–249 CrossRef CAS PubMed.
- N. Pasha, N. Lingaiah, P. S. S. Reddy and P. S. Prasad, Catal. Lett., 2009, 127, 101–106 CrossRef CAS.
- R. Amrousse and T. Katsumi, Catal. Commun., 2012, 26, 194–198 CrossRef CAS PubMed.
- H. Zhou, Z. Huang, C. Sun, F. Qin, D. Xiong, W. Shen and H. Xu, Appl. Catal., B, 2012, 125, 492–498 CrossRef CAS PubMed.
- P. Pomonis, D. Vattis, A. Lycourghiotis and C. Kordulis, J. Chem. Soc., Faraday Trans. 1, 1985, 81, 2043–2051 RSC.
- A. Dandekar and M. A. Vannice, Appl. Catal., B, 1999, 22, 179–200 CrossRef CAS.
- A. K. Ladavos and T. V. Bakas, React. Kinet. Catal. Lett., 2001, 73, 229–235 CrossRef CAS.
- G. Giecko, T. Borowiecki, W. Gac and J. Kruk, Catal. Today, 2008, 137, 403–409 CrossRef CAS PubMed.
- T. Gaidei, N. Pillet, V. Sadov, M. Strukova, S. Filatov, E. Khaustova and N. Yaroshenko, Russ. J. Appl. Chem., 2010, 83, 1154–1158 CrossRef CAS.
- R. S. da Cruz, A. J. S. Mascarenhas and H. M. C. Andrade, Appl. Catal., B, 1998, 18, 223–231 CrossRef CAS.
- R. Joyner and M. Stockenhuber, J. Phys. Chem. B, 1999, 103, 5963–5976 CrossRef CAS.
- L. Li, Q. Shen, J. Yu, J. Li, Z. Hao and Z. P. Xu, Catal. Commun., 2008, 9, 1745–1748 CrossRef CAS PubMed.
- P. K. Roy, R. Prins and G. D. Pirngruber, Appl. Catal., B, 2008, 80, 226–236 CrossRef CAS PubMed.
- A. Ates, A. Reitzmann and G. Waters, Appl. Catal., B, 2012, 119, 329–339 CrossRef PubMed.
- J. Y. Wang, H. A. Xia, X. H. Ju, F. T. Fan, Z. C. Feng and C. Li, Chin. J. Catal., 2013, 34, 876–888 CrossRef CAS.
- K. S. Chang, H. Song, Y. S. Park and J. W. Woo, Appl. Catal., A, 2004, 273, 223–231 CrossRef CAS PubMed.
- L. Obalová, K. Jirátová, F. Kovanda, K. Pacultová, Z. Lacný and Z. Mikulová, Appl. Catal., B, 2005, 60, 289–297 CrossRef PubMed.
- L. Obalová, K. Pacultová, J. Balabánová, K. Jirátová, Z. Bastl, M. Valášková, Z. Lacný and F. Kovanda, Catal. Today, 2007, 119, 233–238 CrossRef PubMed.
- V. K. Tzitzios and V. Georgakilas, Chemosphere, 2005, 59, 887–891 CrossRef CAS PubMed.
- P. Reddy, N. Pasha, M. Chalapathi Rao, N. Lingaiah, I. Suryanarayana and P. Sai Prasad, Catal. Commun., 2007, 8, 1406–1410 CrossRef CAS PubMed.
- H. B. Zhou, P. L. Hu, Z. Huang, F. Qin, W. Shen and H. L. Xu, Ind. Eng. Chem. Res., 2013, 52, 4504–4509 CrossRef CAS.
- N. Russo, D. Mescia, D. Fino, G. Saracco and V. Specchia, Ind. Eng. Chem. Res., 2007, 46, 4226–4231 CrossRef CAS.
- E. Iwanek, K. Krawczyk, J. Petryk, J. W. Sobczak and Z. Kaszkur, Appl. Catal., B, 2011, 106, 416–422 CrossRef CAS PubMed.
- A. Gervasini, P. Carniti and S. Bennici, React. Kinet., Mech. Catal., 2012, 105, 53–67 CrossRef CAS PubMed.
- S. Liu, Y. Cong, C. Kappenstein and T. Zhang, Chin. J. Catal., 2012, 33, 907–913 CrossRef CAS.
- Y. Wu, C. Dujardin, P. Granger, C. Tiseanu, S. Sandu, V. Kuncser and V. I. Parvulescu, J. Phys. Chem. C, 2013, 117, 13989–13999 CAS.
- Z. Lan, S. C. Zhang, L. Q. Wang and C. H. Liu, Ind. Catal., 2002, 10, 54–56 CAS.
- Y. Wang, J. Zhang, J. Zhu, J. Yin and H. Wang, Energy Convers. Manage., 2009, 50, 1304–1307 CrossRef CAS PubMed.
- CELREF unit-cell refinement software for Windows by Laugier and Bochu, http://www.ccp14.ac.uk/.
- A. Heyden, B. Peters, A. T. Bell and F. J. Keil, J. Phys. Chem. B, 2005, 109, 1857–1873 CrossRef CAS PubMed.
- G. D. Pirngruber, P. K. Roy and R. Prins, J. Catal., 2007, 246, 147–157 CrossRef CAS PubMed.
- N. Liu, R. Zhang, B. Chen, Y. Li and Y. Li, J. Catal., 2012, 294, 99–112 CrossRef CAS PubMed.
- Y. Wu, X. Ni, A. Beaurain, C. Dujardin and P. Granger, Appl. Catal., B, 2012, 125, 149–157 CrossRef CAS PubMed.
- R. J. Madon and M. Boudart, Ind. Eng. Chem. Fundam., 1982, 21, 438–447 CAS.
- R. Weber, M. Plinke, Z. T. Xu and M. Wilken, Appl. Catal., B, 2001, 31, 195–207 CrossRef CAS.
- R. Klaewkla, M. Arend and W. F. Hoelderich, Mass Transfer-Advanced Aspects, 2011, pp. 668–684 Search PubMed.
- S. Parres-Esclapez, I. Such-Basañez, M. J. Illán-Gómez, C. Salinas-Martínez de Lecea and A. Bueno-López, J. Catal., 2010, 276, 390–401 CrossRef CAS PubMed.
- A. De Asha, J. Critchley and R. Nix, Surf. Sci., 1998, 405, 201–214 CrossRef CAS.
- E. Wolska and U. Schwertmann, Z. Kristallogr, 1989, 189, 223–237 CrossRef CAS PubMed.
- J. Requies, M. A. Cabrero, V. L. Barrio, M. B. Guemez, J. F. Cambra, P. L. Arias, F. J. Perez-Alonso, M. Ojeda, M. A. Pena and J. L. G. Fierro, Appl. Catal., A, 2005, 289, 214–223 CrossRef CAS PubMed.
- G. Wrobel, M. P. Sohier, A. D'Huysser, J. P. Bonnelle and J. P. Marcq, Appl. Catal., A, 1993, 101, 73–93 CrossRef CAS.
- P. Pietrzyk, F. Zasada, W. Piskorz, A. Kotarba and Z. Sojka, Catal. Today, 2007, 119, 219–227 CrossRef CAS PubMed.
- Z. Zhao, X. Yang and Y. Wu, Appl. Catal., B, 1996, 8, 281–297 CrossRef CAS.
- J. F. Moulder, J. Chastain and R. C. King, Handbook of X-ray photoelectron spectroscopy: a reference book of standard spectra for identification and interpretation of XPS data, Physical Electronics, Eden Prairie, MN, 1995 Search PubMed.
- D. F. Mullica, C. K. C. Lok, H. O. Perkins and V. Young, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 4039–4042 CrossRef CAS.
- C. Suzuki, J. Kawai, M. Takahashi, A.-M. Vlaicu, H. Adachi and T. Mukoyama, Chem. Phys., 2000, 253, 27–40 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS PubMed.
- T. L. Barr, J. Phys. Chem., 1978, 82, 1801–1810 CrossRef CAS.
- A. Y. Dobin, K. Nikolaev, I. Krivorotov, R. Wentzcovitch, E. D. Dahlberg and A. Goldman, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 113408 CrossRef.
| This journal is © The Royal Society of Chemistry 2014 |