DOI:
10.1039/C4RA02373K
(Paper)
RSC Adv., 2014,
4, 19538-19549
A novel pyrene-containing fluorescent organogel derived from a quinoline-based fluorescent porbe: synthesis, sensing properties, and its aggregation behavior†
Received
20th March 2014
, Accepted 15th April 2014
First published on 16th April 2014
Abstract
A new family of pyrene-containing compounds 2–4 derived from aminoquinoline-containing fluorescent probe 1 were successfully synthesized and well characterized. The investigation of the absorption and emission spectra of these compounds revealed that the photophysical properties were significantly affected by the substitution of pyrene. Moreover, these compounds exhibited selective fluorescence behaviour towards Zn2+ in aqueous solution. The gelation properties of these compounds were investigated by the “stable to inversion of a test tube” method. Interestingly, 1,8-bis-substituted compound 4 displayed stable gel-formation properties in acetone, dioxane, tetrahydrofuran, ethyl acetate, chloroform, and dichloromethane. The morphologies of the xerogels were investigated by scanning electron microscopy (SEM) and laser scanning confocal microscopy (LSCM). The concentration- and temperature-dependent emission properties, and concentration- and temperature-variable 1H NMR spectroscopy of compound 4 were investigated, which suggested that both π–π stacking interaction and hydrogen bonding were the driving forces for the process of self-aggregation and the gel formation. In addition, studies on the luminescence properties indicated that 4 had the ability to form a fluorescent organogel with interesting fluorescence behaviour. More importantly, it was found that compound 4 could form a stimuli-responsive gel that had a sensitive gel-to-sol transition response to heating or adding Zn2+.
Introduction
Over the past few decades, supramolecular chemistry1 has witnessed tremendously rapid development in the construction of delicated functional architectures2,3 including smart soft matter.4 Particularly, organogels have attracted a great deal of attention in the areas that range from supramolecular chemistry to materials science because of their unique supramolecular structures and potential applications in template synthesis, drug delivery, and biomimetic systems.5 Generally, organogels are formed through the self-assembly of gelator molecules to the generation of three-dimensional supramolecular network structures with a lot of void spaces occupied by solvent molecules under the suitable conditions. The driving forces for gelator molecules to self-assemble into the entangled nanostructures are generally weak non-covalent interactions such as π–π stacking, hydrogen-bonding, electrostatic interactions, hydrophobic interactions, and van der Waals interactions.
Among various organogels, fluorescent organogels constructed from π-conjugated molecules have drawn significant interest due to their distinctive advantages such as diversity, flexibility, and promising applications in optoelectronics, erasable thermal imaging, and sensing of explosives like trinitrotoluene.6 Moreover, a remarkable variation in the profile of the fluorescence spectrum of a fluorescent organogel may accompany a sol-to-gel or gel-to-sol phase transition process, which may provide the crucial information at a molecular level about the structure changes during the phase transition process. Thus, the incorporation of chromophores makes it easy to predict the gelation phenomenon or explain the structure–property relationship although the gelator–gelator and gelator–solvent interactions are intricate. As a result, a number of fluorescent organogels based on various aromatic building blocks including porphyrins,7 pyrenyl,8 perylene bisimides,9 oligo-(p-phenylenevinylene),10 and naphthalimides11 have been investigated during the recent past years.
On the other hand, the stimuli-responsive organogels are of much interest due to their benefits for the development of sensor devices or applications like drug delivery, controlled release, and catalysis, etc.12 Stimuli-responsive organogels can be rendered sensitive to the external stimulus, such as temperature,13 light irradiation,14 sound,15 pH,16 oxidative/reductive reactions,17 and anions,18 by incorporating a receptor unit or a spectroscopically active unit as part of gelator molecule. For instance, organogels that response to light irradiation have been obtained by incorporating azobenzene, spiropyran, stilbene, etc. into the corresponding organogels.14a,d,17b Similarly, if the organogels contain acidic or basic groups, the gel–sol transition can be reversibly switched by appropriately changing the pH value.19 Notably, even though considerable efforts have been devoted to the development of stimuli-responsive organogels, the related reports are still limited. This might be caused by the fact that the preparation of such organogel needs an effective coupling between the recognition unit and the self-organization unit. Furthermore, it has been recognized that the receptor unit should be an active component participating in the self-assembly process. Therefore, it is still challenging yet desirable to develop stimuli-responsive organogels, especially stimuli-responsive fluorescent organogels.
Herein, in order to construct a stimuli-responsive fluorescent organogel, a new family of pyrene-containing compounds 2–4 were synthesized in this work. Compounds 2–4 were derived from the fluorescent probe 1, which was reported to exhibit characteristic changes in its fluorescence profile in response to Zn2+ through the binding of Zn2+ with three nitrogen atoms (Scheme 1).20 The following principles have been considered in the design of 2–4. Firstly, the long chain, which contains amide and NH groups, not only can act as Zn2+ acceptor but also have the ability to form multiple hydrogen bonds. It's well known that multiple hydrogen bonds may provide driving forces to form organogel in the appropriate solvents.21 Secondly, as a π-conjugated molecule, pyrene and its derivatives have been widely explored in the field of luminescent material.22 With the introduction of ethynyl subunits into the pyrene skeleton, the conjugated fusion of pyrene moiety with quinoline moiety may result in the strong intermolecular π–π stacking, which provides another powerful driving force to form organogel. Thirdly, in order to investigate the influence of the substitution on photophysical properties, sensing properties, and organogel formation, a family of compounds 2–4 with monosubstituted, 1,6-bis-substituted, and 1,8-bis-substituted branches, were designed and synthesized. It was found that 2–4 featured the interesting spectroscopic properties due to the existence of pyrene and quinoline moieties. All of them exhibited good yet different sensing properties to Zn2+. Interestingly, compound 4 presented a good gel formation property in many solvents such as dichloromethane, chloroform, acetone, and tetrahydrofuran, etc. More importantly, it was found that heating or adding Zn2+ could induce the gel–sol phase transition. This work provides an interesting and successful example of constructing stimuli-responsive fluorescent organogel, which obviously enriches the library of stimuli-responsive fluorescent organogels and might be helpful in preparing such type of organogels.
 |
| | Scheme 1 Molecular structures of compounds 2–4. | |
Results and discussion
Synthesis
Compound 1 was synthesized according to the literature procedure.20 The preparation of pyrene-containing compounds 2–4 was outlined in Scheme 2. Iodination of 8-aminoquinoline afforded 5, followed by the reaction with chloroacetyl chloride in room temperature to give compound 6 in high yield (98.2%). Subsequently, a further nucleophile substitution reaction of 6 with 2-(2-aminoethoxy)ethanol was carried out to provide the key intermediate 7. In addition, 1-ethynylpyrene 9, 1,6-diethynylpyrene 14, and 1,8-diethynylpyrene 15 were prepared according to our previous reports.8e It was worth noting that bromination of pyrene resulted in two isomers 1,6-dibromopyrene 10 and 1,8-dibromopyrene 11, which were difficult to be isolated from each other. However, a further alkynylation of 10 and 11 was performed to prepare a mixture of 12 and 13, which could be easily purified with column chromatography to give pure 1,6-diethynyl and 1,8-diethynylpyrene, respectively. Finally, sonogashira coupling reaction of 7 with 9, 14, or 15 generated the series of pyrene-containing compounds 2–4 in acceptable yields, respectively. The molecular structures of compounds 2–4 were characterized by 1H NMR, 13C NMR, and mass spectroscopy, which were in full agreement with the desired structures. It is noteworthy that the 1H NMR spectrum of 4 displayed two singlets at 7.76 and 8.42 ppm, which were assigned to the protons on 4-, 5-, and 9-, 10-substituted location of pyrene, respectively (Fig. S3 in ESI†).21b However, in the 1H NMR spectrum of 3, none of the singlet could be found in the aromatic region. It means that the isomers 3 and 4 could be distinguished from each other base on the singlets.
 |
| | Scheme 2 Synthesis route of target compounds 2–4. | |
Investigation of spectral properties of 2–4
The absorption and emission spectra of compounds 2–4 in acetone (10−5 M) were presented in Fig. 1. The obtained results indicated that the absorption properties were significantly affected by the substitution of pyrene. For example, two absorption bands were observed for 2 at λmax of 391 and 415 nm along with a shoulder peak at 366 nm. Unlike 2, the bisubstituted pyrene derivative 3 had two maximum absorption bands located at 430 and 448 nm, respectively. Moreover, the onset of the absorption of 3 was very broad, which was from 350 to 430 nm. Compound 4, however, which had the same number of substitution groups as 3 but the different substitution position, exhibited two main absorption regions with bands at λmax of 370 and 432 nm, respectively. Furthermore, there were two shoulder peaks located at 410 and 450 nm. It was proposed that the intramolecular charge transfer (ICT) effect induced different UV-Vis absorptions of isomers 3 and 4. Compared to free pyrene (absorption band usually appearing at 320 nm) and 1 (absorption band appearing at 305 nm), compounds 2–4, especially 3 and 4, showed pronounced red-shifted absorption due to the extension of π-conjugation. The additional substituted 8-aminoquinoline group in 3 and 4, as compared to 2, induced a ∼35 nm red shift in the absorption spectrum. Moreover, the broad and loss of vibronic structure absorption spectra of 2–4 indicated that there existed charge transfer between pyrene and 8-aminoquinoline.
 |
| | Fig. 1 UV-Vis (a) and fluorescence emission (b) spectra of 2–4 (10 μM) in acetone. | |
The UV-Vis absorption of 2–4 in different solvents including dichloromethane, ethyl acetate, tetrahydrofuran, dimethylbenzene, dioxane, toluene, chloroform, n-propanol, tetrahydrofuran–water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), acetone–water (1:1, v/v), dioxane–water (1:1, v/v), and n-propanol–water (1:1, v/v) were studied. As shown in Fig. S7–S9a,† the shapes of the absorption spectra of 2–4 showed almost no change in various solvents. Meanwhile, a slight shift in wavelength and a moderate change of extinction coefficient of 2–4 in different solvents were observed. The absorption spectra shifts may arise from either the physical intermolecular solute–solvent interactions (e.g. hydrogen-bonding interaction) or the chemical processes such as charge transfer or proton transfer, etc.23 Moreover, a relative higher molar extinction coefficient in the tested solvents indicated the more effective π → π* and n → π* transition for the absorption bands in the π-conjugated molecules. Therefore, the results disclosed that the solvents featured the moderate effect on UV absorption spectra of 2–4.
:1, v/v), acetone–water (1:1, v/v), dioxane–water (1:1, v/v), and n-propanol–water (1:1, v/v) were studied. As shown in Fig. S7–S9a,† the shapes of the absorption spectra of 2–4 showed almost no change in various solvents. Meanwhile, a slight shift in wavelength and a moderate change of extinction coefficient of 2–4 in different solvents were observed. The absorption spectra shifts may arise from either the physical intermolecular solute–solvent interactions (e.g. hydrogen-bonding interaction) or the chemical processes such as charge transfer or proton transfer, etc.23 Moreover, a relative higher molar extinction coefficient in the tested solvents indicated the more effective π → π* and n → π* transition for the absorption bands in the π-conjugated molecules. Therefore, the results disclosed that the solvents featured the moderate effect on UV absorption spectra of 2–4.
Compared to the absorption spectra, the emission spectra of compounds 2–4 exhibited the slighter dependence on the substitution of pyrene in acetone (Fig. 1b). For instance, compounds 3 and 4 showed the similar emission spectral shapes and exhibited two main emission regions with bands about at 465 and 490 nm in acetone. The fluorescence emission spectra for 2–4 were also measured in various solvents for certifying the influence of solvent on their emission (Fig. S7–S9b in ESI†). The results indicated that fluorescence emission of compound 2 was more sensitive to solvents than that of 3 and 4. For example, in different tested solvents, the emission spectra of compounds 3 and 4 showed the similar shapes and a slight wavelength shift. However, compound 2 displayed totally different emission spectra in non-protonic solvents and protonic solvents. In the non-protonic solvents such as dioxane, ethyl acetate, and tetrahydrofuran, compound 2 showed two main emission band peaks at 436 and 460 nm, which was the characteristic feature of monomer of pyrene derivatives. In comparison, however, compound 2 exhibited the excimer peak at the longer wavelength regions in the protonic solvents such as n-propanol, tetrahydrofuran–water, acetone–water, dioxane–water, and n-propanol–water. This observation might be caused by the fact that the bisubstituted 3 and 4, as compared with monosubstituted 2, have the larger steric hindrance, thus preventing the formation of excimer in protonic solvents.
Metal ions sensing properties of 2–4
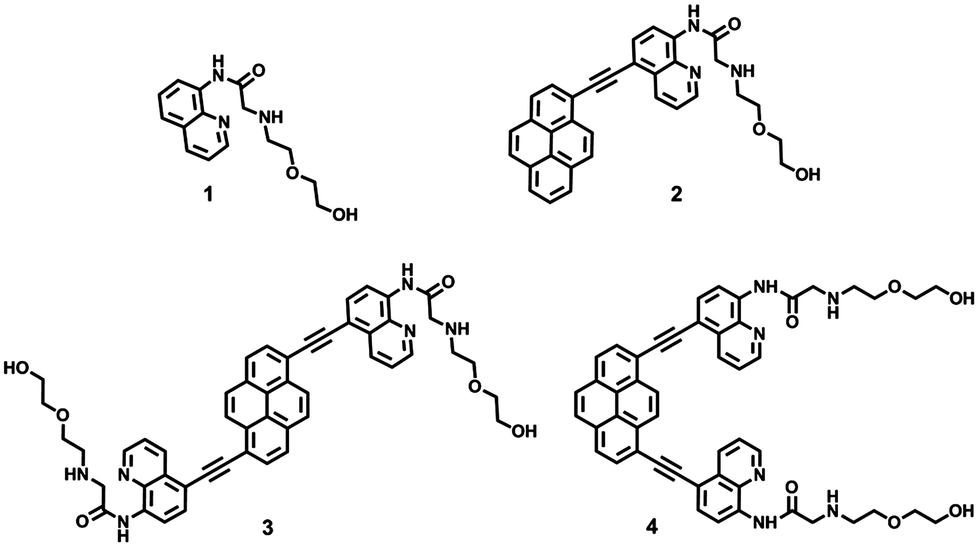
In order to evaluate the selectivity of 2–4, various metal ions such as Zn2+, Mn2+, Ba2+, Hg2+, Ni2+, Cu2+, Co2+, Pb2+, Mg2+, Cd2+, Fe2+, Al3+, Ag+, Li+, and Na+ were employed. As shown in Fig. 2, upon adding various metal ions to the solution of 2, 3, and 4, respectively, only Zn2+ induced a remarkable red-shift of emission spectra by comparison with that of free 2–4 in the solution. For example, with the addition of increasing amounts of Zn2+ to 2, a slight decrease in fluorescent intensity and a 38 nm red-shift from 480 to 518 nm of fluorescence emission were observed. According to the previous report, it is speculated that the red-shift of emission of 2 upon adding Zn2+ was attributed to the deprotonation of amide when binding with Zn2+.20 Though a significant fluorescence quenching was detected with the addition of Cu2+, Co2+, Fe2+, and Pb2+ due to their well-known paramagnetic effect,24 it should be noted that the fluorescence emission wavelength did not change. As shown in Fig. S10–S12 in ESI†, about 2-fold increase in the fluorescence intensity ratio of 2 at 518 and 480 nm (I546nm/I465nm) was observed following the addition of 4 equiv. Zn2+. Under the identical conditions, Cu2+ induced 50% increase in the fluorescence intensity ratio at 518 to 480 nm (I518nm/I480nm). More interestingly, a remarkable red-shift of 81 nm was observed for 3 with the addition of 4 equiv. Zn2+, which induced about 41-fold increase in the fluorescence intensity ratio of 3 at 546 and 465 nm (I518nm/I480nm). Similarly, the addition of Zn2+ to the solution of 4 resulted in a 57 nm red-shift from 466 to 523 nm of fluorescence emission and 14-fold increase in the fluorescence intensity ratio value at 523 and 466 nm (I523nm/I466nm). However, under the identical conditions, Cu2+ induced a 1.8-fold increasing of fluorescence intensity ratio at 523 to 466 nm, ca. 12% compared to Zn2+ (Fig. 3). The other ions produced minor changes in the fluorescence intensity ratio value. From the view of the application of fluorescent probe, 2, 3 and 4 did not exhibit better behavior than that of 1 due to the large overlap of fluorescence emission spectra between free compound (2, 3, and 4) and the corresponding complex (4 equiv. Zn2+ was added to the solution of 2, 3, and 4, respectively), however, all of them displayed selective fluorescence behaviour towards Zn2+ in aqueous solution.
 |
| | Fig. 2 Fluorescence emission spectra of 2 (a, 10 μM), 3 (b, 10 μM), and 4 (c, 10 μM) in the presence of 40 μM of Zn2+ in acetone–water (1:1, v/v, 10 mM Tris–HCl, pH 7.2). | |
 |
| | Fig. 3 Fluorescence intensity ratio of 4 (10 μM) at 523 and 466 nm (I523nm/I466nm) in the presence of 4 equiv. various metal ions to free 4 (10 μM) at 523 and 466 nm (I523nm/I466nm) in acetone–water (1:1, v/v, 10 mM Tris–HCl, pH 7.2). | |
Gelation properties
The gelation properties of compounds 1–4 were investigated by the “stable to inversion of a test tube” method in various polar or nonpolar, aromatic or nonaromatic solvents such as n-heptane, acetone, dioxane, tetrahydrofuran, ethyl acetate, toluene, dimethylbenzene, n-propanol, chloroform, dichloromethane, n-propanol, tetrahydrofuran–water (1:1, v/v), acetone–water (1:1, v/v), dioxane–water (1:1, v/v), and n-propanol–water (1:1, v/v). The results were summarized in Table 1. It was surprised to find that compound 4 exhibited the stable gel-formation property in acetone, dioxane, tetrahydrofuran, ethyl acetate, chloroform, and dichloromethane (Fig. 4). While compounds 1–3 could not gel in the tested solvents. For instance, compound 2 was insoluble in all of the tested solvents both at room temperature and at the boiling point of the tested solvents. Similarly, compound 3 was insoluble in n-heptane, acetone, tetrahydrofuran, ethyl acetate, toluene, and dimethylbenzene both at room temperature and at the boiling point of the tested solvents. Although compound 3 could dissolve in dioxane, n-propanol, chloroform, and dichloromethane, it precipitated gradually out of these solvents upon subsequently cooling to the room temperature. The results revealed that the structural effects, such as the number of substitution groups and the substitution position have an observable influence on the gel formation.
Table 1 Gel properties of 1–4 in organic solventsa
| Solvent |
1 |
2 |
3 |
4 |
| G = gel; I = insoluble; PG = partial gel; P = precipitation; S = solution. The values in parentheses are the CGC in mg mL−1. |
| n-Heptane |
I |
I |
I |
I |
| Acetone |
S |
I |
I |
G(8.24)b |
| Dioxane |
S |
I |
P |
G(13.73) |
| Tetrahydrofuran |
S |
I |
I |
G(13.73) |
| Ethyl acetate |
S |
I |
I |
G(13.73) |
| Toluene |
S |
I |
I |
I |
| Dimethylbenzene |
S |
I |
I |
I |
| n-Propanol |
S |
I |
P |
P |
| Chloroform |
S |
I |
P |
G(8.24) |
| Dichloromethane |
S |
I |
P |
G(8.24) |
| Tetrahydrofuran–water |
S |
I |
I |
I |
| Acetone–water |
S |
I |
I |
I |
| Dioxane–water |
S |
I |
I |
PG |
| n-Propanol–water |
S |
I |
I |
I |
 |
| | Fig. 4 The gel state photographs (a, under ambient; b, irradiated at 365 nm by a handheld UV lamp) of 4 in various solvents: (1) acetone; (2) dioxane; (3) tetrahydrofuran; (4) ethyl acetate; (5) chloroform; (6) dichloromethane. | |
The critical gelation concentrations (CGC) of 4 in acetone, chloroform, and dichloromethane was 8.24 mg mL−1, indicating that one molecule of 4 could entrap approximately 1377, 1256, and 1562 solvent molecules, respectively. Moreover, the CGC of 4 in dioxane, tetrahydrofuran, and ethyl acetate was determined to be 13.73 mg mL−1. These data demonstrated that 4 formed LMOGs more easily in acetone, chloroform, and dichloromethane than that in dioxane, tetrahydrofuran, and ethyl acetate.
To investigate the morphologies of the obtained organogels, scanning electron microscopy (SEM) was performed with the corresponding xerogels (air-dried gels). Gel samples were slowly evaporated to dryness, and then the resultant xerogels were examined (Fig. 5 and 6 and Fig. S13–S17 in ESI†). As shown in Fig. 5, the morphologies of xerogels were dependent on the solvents of gelation. For example, the xerogels of 4 from polar solvents such as acetone, dioxane, and tetrahydrofuran exhibited twining agglomerates, while cross-linked massive morphology was found in the case of ethyl acetate. The solvent dependent morphology of gel 4 may be due to the dipolar structure of 4 and the competition interactions between gelator–gelator and solvent–gelator. Moreover, for the xerogel from compound 4 in nonpolar solvents such as chloroform and dichloromethane, it appeared as a 3D network comprised of entangled fibers with diameter of 200–600 nm.
 |
| | Fig. 5 SEM images of the xerogels of 4 in (a) acetone; (b) dioxane; (c) tetrahydrofuran; (d) ethyl acetate; (e) chloroform; (f) dichloromethane. Scale bars: 5.0 μm. | |
 |
| | Fig. 6 SEM images of the xerogels of 4 in chloroform. Scale bars: (a) 1.0 μm; (b) 2.0 μm; (c) 5.0 μm; (d) 10.0 μm. | |
Study of the aggregation process in solution
With the aim to obtaining the insight into the aggregation mode during the gel formation, a further spectroscopic investigation of these gelators was carried out. It should be noted that the 4 showed concentration- and temperature-dependent emission properties (Fig. 7a and b). For example, upon increasing the concentration from 1.0 × 10−6 to 1.0 × 10−3 M at 298 K, a broad structureless band at 507 nm was observed, accompanied by the disappearance of the emission bands at 466 and 494 nm, which indicated the formation of a pyrene excimer. Meanwhile, as shown in Fig. 7b, upon warming a gelled solution of 4 from 298 to 328 K, the emission spectra of 4 featured a slight blueshift (ca. 10 nm). Furthermore, when the solution was heated at 328 K, there appeared a new peak at 500 nm. Over the same temperature range the sample undergoes a gel–sol transition. Therefore, the thermally induced shift in the emission spectra was clearly associated with the phase transition.
 |
| | Fig. 7 Emission spectra of 4 in acetone (a) at different concentrations and (b) at different temperatures (CGC concentration). | |
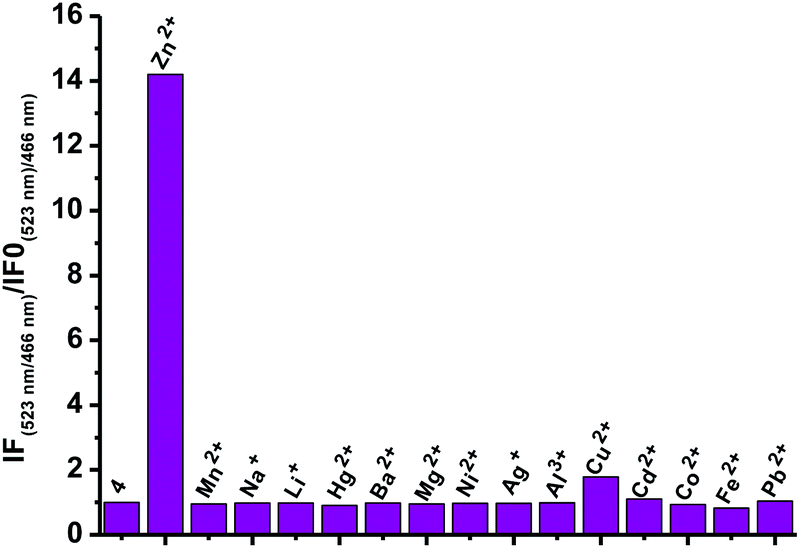
NMR spectra can not only reveal the changes in hydrogen bonding, but also disclose the interactions between aromatic moieties.25 Therefore, the concentration- and temperature-dependent 1H NMR spectroscopic studies of 4 were performed to probe the driving forces for the self-assembly process (Fig. 8 and 9). The 1H NMR spectra displayed distinctly concentration- and temperature-dependent features. For instance, as the concentration increased, the resonance signals corresponding to the aromatic protons displayed an obvious upfield (e.g., H9,10 and H4,5, upfielded from 8.84 and 8.09 ppm to 8.39 and 7.73 ppm, respectively). Similarly, the aromatic protons showed an obvious upfield shift upon decreasing temperature (e.g., H9,10 and H4,5, upfielded from 8.46 and 7.79 ppm to 8.25 and 7.61 ppm, respectively). It is noteworthy that, even in the gelling solvent (CDCl3), the aromatic protons showed upfield shift when the sample was cooled from the hot solution state to the gel state (Fig. 10). It is well known that the upfield shift of the aromatic proton resonance of a π-conjugated system reflects an increase in the π–π stacking interaction.8g Thus, the extent of the π–π stacking interaction in 4 increased with the increase of concentration and the decrease of temperature.
 |
| | Fig. 8 Partial 1H NMR spectra (400 MHz, CD2Cl2–CD3OD = 3:2, 298 K) of compound 4 at different concentrations. | |
 |
| | Fig. 9 Partial 1H NMR spectra (400 MHz, CD2Cl2–CD3OD = 3:2) of compound 4 at different temperature. | |
 |
| | Fig. 10 Partial 1H NMR spectra (400 MHz, CDCl3) of compound 4 at different temperature. | |
Moreover, it was found that the NH resonance displayed an obvious upfield shift upon the increase of temperature, which was indicative of the existence of hydrogen-bonding interactions during the formation of supramolecular aggregate in the case of 4. Temperature dependence of NH chemical shift (Δδ/ΔT) has been widely used as a tool for the study of intramolecular and intermolecular H-bonding during the assembling process for the hydrogen bonded gelator molecules.6f A small (Δδ/ΔT) value for the NH resonance indicated the participation of NH proton in an internal H-bond or it may also be interpreted as an unbound NH that was completely exposed to the solvent. On the other hand, relatively larger temperature coefficient in solvent showed the involvement of amide NHs in intermolecular H-bonding. VT-NMR studies (Fig. 11) conducted with compound 4 over the temperature range 293–323 K showed that there was a steady upfield chemical shift of NH protons upon increasing temperature. Moreover, the moderate coefficient (Δδ/ΔT) in ppb/K = (1.37 ppb/T) indicated a moderate intermolecular hydrogen bonding in self-assembled state responsible for gelation. Therefore, concentration- and temperature-1H NMR spectroscopic studies suggested that both π–π stacking interaction and hydrogen bonding were the driving forces for the process of self-assembly and gelation of the gelator 4.
 |
| | Fig. 11 Chemical shift of NH resonance (ppm) of 4 at different temperature in CDCl3. | |
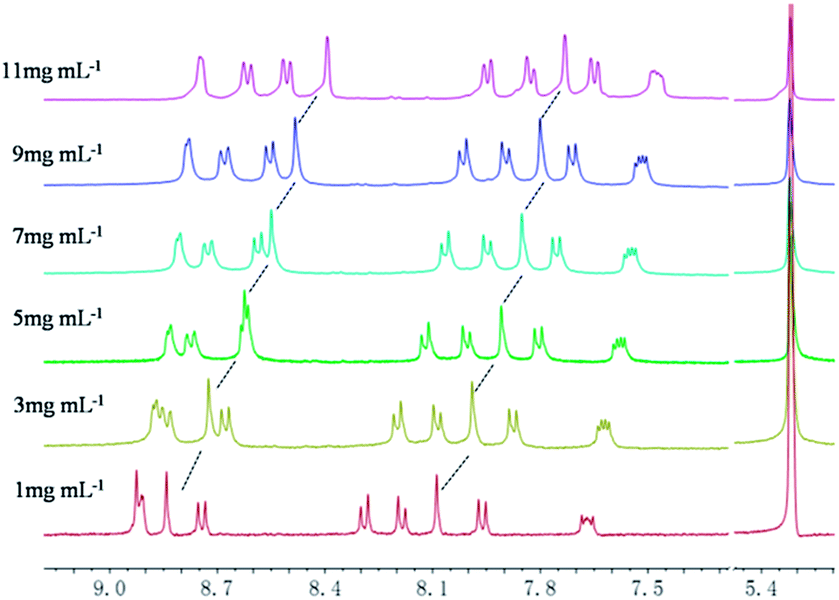
Based on all above experimental facts, a possible mechanism for the formation of such LMOG was proposed (Fig. 12). Initially, the primary-ordered aggregates were formed from the simple building block mostly driven by π–π stacking interaction, hydrogen-bonding, and other possible non-covalent interactions. Subsequently, these primary aggregates evolved to be the secondary fiber-like structures or twining agglomerates, which ultimately intertwine to form large intense three-dimensional networks or other morphologies with large bulks. Although it is still very difficult to demonstrate the definite process of the formation of such organogel, there is no doubt that π–π stacking interaction and hydrogen-bonding played an important role in the formation of the ordered aggregates.
 |
| | Fig. 12 Schematic illustration for the formation of gels from 4 in tested solvents. | |
Emission investigation of fluorescent organogel
As mentioned previously, the fluorescent organogels containing π-conjugated chromophores have received considerable attention because of their potential applications in optoelectronics, erasable thermal imaging, and sensing of explosives.6 Inspired by the existence of a large π-conjugated system in compound 4, the further spectroscopic investigation on this new fluorescent organogel was carried out. As shown in Fig. 13, compound 4 showed two shoulder peaks located at 468 and 495 nm in solution, which arised from the non-aggregated of pyrene-containing compound. Under self-assembly conditions, in the gel state, 4 exhibited a excimer emission peak centered at 546 nm. This indicated that pyrenyl group of this gelator 4 was dimerized in the gel state, which might be caused by the presence of strong π–π stacking interaction. The observed significant red shift of fluorescence emission maxima was obvious due to the presence of a strong molecular aggregation in the gel state. Thus, the fluorescence emission investigation of 4 provided further evidence that π–π stacking interaction contributed to the formation of organogel. Moreover, as shown in Fig. 4b, upon irradiation at 365 nm by a handheld UV lamp, compound 4 exhibited stable and chartreuse fluorescence gels in various solvents such as acetone, dioxane, tetrahydrofuran, ethyl acetate, chloroform and dichloromethane. With the aim to obtaining further insight into the morphologies and optical properties of this new LMOG, the laser scanning confocal microscopy (LSCM) was performed on the xerogels of 4. Notably, the green luminescence was emitted from a nanoscale structure, which was directly observed by LSCM images as shown in Fig. 14. Moreover, the investigation of LSCM on samples of 4 offered similar morphologies as that observed in the SEM studies. The 3-D networks comprised by fibers structures were found in LSCM images of 4. The results indicated that 4 had the ability to form fluorescent organogel with good fluorescence behaviour.
 |
| | Fig. 13 Nomalized fluorescence emission spectra of compound 4 in solution (10−5 M) and in the gel state (CGC concentration) in acetone. | |
 |
| | Fig. 14 LCSM images (a and d) and bright-field images (b and e) of xerogels of compound 4 in chloroform. (c) Was the overlap of (a) and (b). (f) Was the overlap of (d and e). λex = 405 nm, emission was collected at 450–550 nm. | |
Heating or Zn(II)-induced gel–sol phase transition
Similar to the reported gels,13 the gel–sol interconversion of 4 was fully thermoreversible by several cycles of heating and cooling. As shown in Fig. 15, the gel was found to be stable in the room temperature, however, when the temperature of gel was heated to the boiling point of the solvent, a clear and yellow solution was observed. Subsequently, when the hot solution of 4 was cooled down to the room temperature, a stable gel re-formed. The gel returned to a yellow solution after re-heating. The cycles of gel-to-sol transition upon heating and sol-to-gel transition upon cooling were reversible.
 |
| | Fig. 15 Photographs demonstrating of the cycles of gel–sol transition of 4 in acetone by heating and cooling and the gel–sol transition of 4 in acetone by addition of Zn2+. | |
As discussed above, compound 4 was able to bind with Zn2+ through the binding of NH group with Zn2+. Thus, the intermolecular H-bonding between the NH groups would be disrupted in the presence of Zn2+. As shown in Fig. 15, the addition of 0.5 equiv. Zn2+ to the gel of 4 induced an apparent and fast gel-to-sol transition within 30 seconds. This observation revealed that the addition of Zn2+ destroyed the H-bonding, one of the driving forces for the gel formation, thus leading to the fast gel-to-sol transition. The results proved that compound 4 could form a stimuli-responsive gel that characterized a sensitive gel-to-sol transition response to Zn2+. Moreover, the solution of 4 containing Zn2+ could not be transformed into the gel under heating and cooling, which also provided support for the assumption that the intermolcular H-bonds between the NH groups should be responsible for the gel formation.
With the aim to obtaining the further insight into the stimuli-responsive properties of gelator 4, the investigations of the responsive behavior of organogels 4 to Zn2+ with different concentrations and in different solutions have been carried out. As shown in Fig. S18a,† the addition of 0.1 equiv. Zn2+ to the gel of 4 in acetone induced the partial gel-to-sol transition within 10 minutes. However, the addition of 0.2 equiv. Zn2+ to the gel of 4 in acetone induced an apparent and fast gel-to-sol transition within 1 minute. The results proved that the gel of 4 exhibited highly sensitive gel-to-sol transition response to Zn2+ in acetone.
Moreover, the solvent influence on the stimuli-responsive property of 4 was discussed. As shown in Fig. S18a,† the addition of 0.4 equiv. Zn2+ to the gel of 4 in dioxane induced the partial gel-to-sol transition within 30 minutes. The addition of Zn2+ up to 0.5 equiv. was able to induce total gel-to-sol transition within 15 minutes. The results indicated that the gel of 4 was more stable in dioxane than that in acetone, which might be attributed to the fact that dioxane has a higher viscosity than acetone.
In order to give a better understand of the application of the gel, the fluorescent change of the gel with the addition of Zn2+ has been carried out. For example, as shown in Fig. 16, the compound 4 showed an emission band peak at 546 nm in the gel state (red curve). However, the addition of 0.5 equiv. Zn2+ to the gel of 4 induced a gel-to-sol transition accompanied with a 7 nm blue-shift from 546 nm to 539 nm (black curve) of the fluorescence emission. The blue-shift of emission provided the further evidence that a weaker intermolecular aggregation of compound 4 existed in the solution state than that in the gel state.
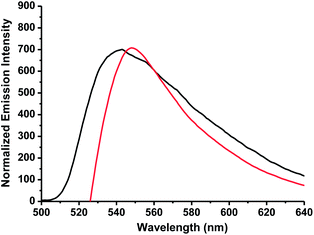 |
| | Fig. 16 Normalized fluorescence emission spectra of compound 4 in the gel state (CGC concentration) absence of Zn2+ (red curve) and in the presence of 0.5 equiv. Zn2+ (black curve) in acetone. | |
Conclusions
By utilizing the sonogashira coupling reaction of ethynylpyrene with 5-I-aminoquinoline derivatives as a key step, a new family of pyrene-containing compounds 2–4 derived from fluorescent probe 1 were successfully synthesized. The molecular structures of these new compounds were fully characterized by using multiple nuclear NMR (1H and 13C) and mass spectrometry. It was found that compounds 2–4 showed pronounced red-shifted absorption and emission compared to free pyrene and 1. Moreover, the absorption and emission properties of these compounds were significantly affected by the substitution of pyrene, such as the numbers and the position of substituent. Furthermore, these compounds exhibited the selective fluorescence behaviour towards Zn2+ in aqueous solution.
It was interesting to find that compounds 2–4 displayed unexpectedly different gelation behaviour, for example, only 1,8-bis-substituted compound 4 exhibited the stable gel-formation property in acetone, dioxane, tetrahydrofuran, ethyl acetate, chloroform, and dichloromethane. The morphologies of the xerogels were investigated by scanning electron microscopy (SEM) and laser scanning confocal microscopy (LSCM). The concentration- and temperature-dependent emission properties, and concentration- and temperature-dependent 1H NMR spectroscopic of 4 revealed that both π–π stacking interaction and hydrogen bonding were the driving forces for the process of self-aggregation and the gel formation. In addition, the studies on luminescence properties indicated that 4 had the ability to form fluorescent organogel with good fluorescence behaviour. More importantly, it was found that that compound 4 could form a stimuli-responsive gel that characterized a sensitive gel-to-sol transition response to heating or Zn2+.
In summary, we have designed and synthesized a family of ethynylpyrene derivatives, which presented interesting luminescence properties and different gel formation properties. More importantly, this work provide an interesting and successful example of constructing stimuli-responsive fluorescent organogel derived from an aminoquinoline-based fluorescent Zn2+ probe with an alkoxyethylamino chain as receptor. This research obviously enriches the library of stimuli-responsive fluorescent organogels and might be helpful in preparing such type of organogels.
Experimental section
All reagents and solvents were purchased from commercial sources. THF was distilled from sodium, Et3N and Et2NH were dried from potassium hydroxide; both solvents were degassed under N2 for 30 min before use. All reactions were performed in standard glassware under an inert N2 atmosphere. Compounds 5, 9, 14, and 15 were prepared as the reported procedure. Unless specifically mentioned, 1H NMR and 13C NMR spectra were recorded on Bruker 400 MHz NMR Spectrometer at 298 K. The 1H and 13C NMR chemical shifts were reported relative to residual solvent signals. Coupling constants (J) are denoted in Hz and chemical shifts (δ) in ppm. Multiplicities were denoted as follows: s = singlet, d = doublet, m = multiplet.
The metal salts used were Fe(ClO4)2, Zn(ClO4)2·6H2O, Co(ClO4)2·6H2O, Ni(ClO4)2·6H2O, Ba(ClO4)2·3H2O, Pb(ClO4)2·3H2O, Cd(ClO4)3·6H2O, Cu(ClO4)2·6H2O, Mn(ClO4)2·6H2O, LiClO4·3H2O, NaClO4·H2O, AgClO4·H2O, Hg(ClO4)2·3H2O, Al(ClO4)3·9H2O, and Cu(ClO4)2·6H2O.
UV-Vis and fluorescence measurements
UV/Vis spectra were recorded on a Cary 50Bio UV/Visible spectrophotometer. Emission spectra were measured on a Cary Eclipse fluorescence spectrophotometer. Samples for absorption and emission measurements were contained in 1 cm × 1 cm quartz cuvette.
Gelation test
A known weight of the compound to be tested and a measured volume of selected pure solvent were placed into a sealed test tube and the system was heated in a water bath until the solid was dissolved, then, the solution was cooled to room temperature in air, and finally the test tube was inversed to look at if the solution inside could still flow. A positive test is obtained if the flow test is negative. When a gel was formed at this stage, it was denoted as “G”. For systems, in which only solution was remained until the end of the tests, they were referred to as “S” (solution). When the gelator of a system appeared as precipitate, the system was denoted as “P” (precipitation). The system, in which the potential gelator could not be dissolved even at the boiling point of the solvent, was called an insoluble system (I).
SEM measurements
SEM images of the xerogels were obtained using an S-4800 (Hitachi Ltd) with an accelerating voltage of 1.0 kV or 10.0 kV. Samples were prepared by dropping dilute gels onto a silicon wafer.
Synthesis of compound 2
A 250 mL Schlenk flask was charged with compounds 7 (2 mmol, 830.5 mg), 9 (1 mmol, 226.3 mg) and cuprous iodide (0.05 mmol, 9.5 mg), degassed, and back-filled three times with N2. Triethylamine (70 mL) and dried THF (70 mL) were introduced into the reaction flask by syringe. The reaction was stirred under an inert atmosphere at 75 °C for 12 h. The solvent was removed by evaporation on a rotary evaporator. The residue was purified by column chromatography on silica gel (dichloromethane–methanol, 99:1) to afford 2 as yellow solid (37.3%). 1H NMR (DMSO-d6, 400 MHz) δ: 11.34 (s, 1H), 9.09 (s, 1H), 8.97 (d, J = 8.4 Hz, 1H), 8.79 (d, J = 8.0 Hz, 1H), 8.71 (d, J = 9.2 Hz, 1H), 8.41 (m, 6H), 8.27 (q, J = 8.8 Hz, 2H), 8.16 (d, J = 8.0 Hz, 2H), 7.90 (q, J = 4.0 Hz, 1H), 3.89 (s, 2H), 3.68 (s, 2H), 3.53 (d, J = 4.4 Hz, 2H), 3.48 (d, J = 4.4 Hz, 4H), 3.04 (s, 2H). 13C NMR (DMSO-d6, 100 MHz) δ: 149.8, 138.0, 135.1, 134.7, 132.0, 131.1, 130.9, 130.8, 130.5, 129.9, 129.0, 128.5, 127.8, 127.2, 126.8, 126.1, 126.0, 125.0, 124.8, 123.7, 123.5, 123.4, 116.6, 116.1, 114.3, 93.1, 92.1, 72.2, 67.9, 60.1, 51.2, 47.8.
Synthesis of compound 3
A 250 mL Schlenk flask was charged with compounds 7 (2.16 mmol, 895.8 mg), 14 (0.719 mmol, 180.0 mg) and cuprous iodide (0.07 mmol, 13.7 mg), degassed, and back-filled three times with N2. Triethylamine (50 mL) and dried THF (50 mL) were introduced into the reaction flask by syringe. The reaction was stirred under an inert atmosphere at 75 °C for 12 h. The solvent was removed by evaporation on a rotary evaporator. The residue was purified by column chromatography on silica gel (dichloromethane–methanol, 99:1) to afford 3 as red solid (38.9%). 1H NMR (DMSO-d6, 400 MHz) δ: 11.53 (s, 2H), 9.00 (d, J = 2.8 Hz, 2H), 8.85 (d, J = 8.4 Hz, 2H), 8.72 (d, J = 8.0 Hz, 2H), 8.61 (d, J = 8.8 Hz, 2H), 8.37 (d, J = 8.0 Hz, 2H), 8.34–8.26 (m, 4H), 8.01 (d, J = 8.0 Hz, 2H), 7.82 (q, J = 4.0 Hz, 2H), 3.62 (t, J = 7.2, 10.4 Hz, 4H), 3.52–3.47 (m, 8H), 3.45 (d, J = 3.2 Hz, 4H), 2.83 (t, J = 4.8, 10.0 Hz, 4H). 13C NMR (DMSO-d6, 100 MHz) δ: 170.9, 149.7, 137.8, 135.2, 134.6, 132.1, 131.0, 130.8, 130.3, 128.7, 127.7, 125.8, 123.4, 117.7, 115.1, 113.5, 92.7, 72.2, 72.1, 70.0, 66.4, 60.2, 60.1, 53.2, 48.9. HRMS m/z: (M + H)+ calcd for C50H45N6O6, 825.3401; found, 825.3422.
Synthesis of compound 4
A 250 mL Schlenk flask was charged with compounds 7 (1.8 mmol, 750 mg), 15 (0.799 mmol, 200.0 mg) and cuprous iodide (0.06 mmol, 12.2 mg), degassed, and back-filled three times with N2. Triethylamine (60 mL) and dried THF (60 mL) were introduced into the reaction flask by syringe. The reaction was stirred under an inert atmosphere at 75 °C for 12 h. The solvent was removed by evaporation on a rotary evaporator. The residue was purified by column chromatography on silica gel (dichloromethane–methanol, 99:1) to afford 4 as red solid (51.1%). 1H NMR (CD2Cl2–CD3OD = 3:2, 400 MHz) δ: 8.76 (d, J = 4.0 Hz, 2H), 8.64 (d, J = 8.4 Hz, 2H), 8.53 (d, J = 8.0 Hz, 2H), 8.42 (s, 2H), 7.97 (d, J = 7.6 Hz, 2H), 7.85 (d, J = 8.0 Hz, 2H), 7.76 (s, 2H), 7.68 (d, J = 8.0 Hz, 2H), 7.49 (q, J = 4.4 Hz, 2H), 3.70 (dd, J = 9.2, 4.8 Hz, 8H), 3.60–3.54 (m, 8H), 2.92 (t, J = 9.6, 4.4 Hz, 4H). 13C NMR (DMSO-d6, 100 MHz) δ: 171.2, 149.7, 137.8, 135.3, 134.6, 132.1, 131.1, 130.8, 130.3, 128.2, 127.7, 126.4, 125.8, 123.4, 117.6, 115.0, 113.5, 92.9, 92.7, 72.2, 70.2, 60.2, 53.4, 49.0. HRMS m/z: (M + H)+ calcd for C50H45N6O6, 825.3401; found, 825.3416.
Acknowledgements
The work was financially supported by the National Natural Science Foundation of China (no. 21302058), the Research Fund for the Doctoral Program of Higher Education of China (no. 20130076120006), the Opening Project of Shanghai Key Laboratory of Chemical Biology, and the Fundamental Research Funds for the Central Universities. We also appreciate valuable discussion with Prof. Hai-Bo Yang (East China Normal University).
Notes and references
-
(a) C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 7017 CrossRef CAS;
(b) J.-M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, Wiley-VCH, Weinheim, Germany, 1995 Search PubMed;
(c) D. J. Cram and J. M. Cram, Container Molecules and Their Guests, Royal Society of Chemistry, Cambridge, UK, 1994 Search PubMed;
(d) J. W. Steed and J. L. Atwood, Supramolecular Chemistry, Wiley and Sons, New York, 2000 Search PubMed.
-
(a) D. Fielder, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 349 CrossRef PubMed;
(b) N. C. Gianneschi, M. S. Maser III and C. A. Mirkin, Acc. Chem. Res., 2005, 38, 825 CrossRef CAS PubMed;
(c) Q.-F. Sun, J. Iwasa, D. Ogawa, Y. Ishido, S. Sato, T. Ozeki, Y. Sei, K. Yamaguchi and M. Fujita, Science, 2010, 328, 1144 CrossRef CAS PubMed;
(d) F. Lin, H.-Y. Peng, J.-X. Chen, D. T. W. Chik, Z. Cai, K. M. C. Wong, V. W. W. Yam and H. N. C. Wong, J. Am. Chem. Soc., 2010, 132, 16383 CrossRef CAS PubMed;
(e) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed;
(f) R. S. Forgan, J.-P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434 CrossRef CAS PubMed;
(g) M.-X. Wang, Acc. Chem. Res., 2012, 45, 182 CrossRef CAS PubMed.
-
(a) B. H. Northrop, H.-B. Yang and P. J. Stang, Inorg. Chem., 2008, 47, 11257 CrossRef CAS PubMed;
(b) H.-B. Yang, B. H. Northrop, Y.-R. Zheng, K. Ghosh and P. J. Stang, J. Org. Chem., 2009, 74, 7067 CrossRef CAS PubMed;
(c) H.-B. Yang, B. H. Northrop, Y.-R. Zheng, K. Ghosh, M. M. Lyndon, D. C. Muddiman and P. J. Stang, J. Org. Chem., 2009, 74, 3524 CrossRef CAS PubMed;
(d) Y.-R. Zheng, H.-B. Yang, K. Ghosh, L. Zhao and P. J. Stang, Chem.–Eur. J., 2009, 15, 7203 CrossRef CAS PubMed;
(e) G.-Z. Zhao, L.-J. Chen, C.-H. Wang, H.-B. Yang, K. Ghosh, Y.-R. Zheng, M. M. Lyndon, D. C. Muddiman and P. J. Stang, Organometallics, 2010, 29, 6137 CrossRef CAS;
(f) X.-D. Xu, H.-B. Yang, Y.-R. Zheng, K. Ghosh, M. M. Lyndon, D. C. Muddiman and P. J. Stang, J. Org. Chem., 2010, 75, 7373 CrossRef CAS PubMed;
(g) Y.-R. Zheng, K. Ghosh, H.-B. Yang and P. J. Stang, Inorg. Chem., 2010, 49, 4747 CrossRef CAS PubMed;
(h) Q. Han, Q.-J. Li, L.-L. Wang, J. He, H. Tan, Z. Abliz, C.-H. Wang and H.-B. Yang, J. Org. Chem., 2011, 76, 9660 CrossRef CAS PubMed;
(i) G.-Z. Zhao, Q.-J. Li, L.-J. Chen, H. Tan, C.-H. Wang, D.-X. Wang and H.-B. Yang, Organometallics, 2011, 30, 5141 CrossRef CAS;
(j) G.-Z. Zhao, Q.-J. Li, L.-J. Chen, H. Tan, C.-H. Wang, D. A. Lehman, D. C. Muddiman and H.-B. Yang, Organometallics, 2011, 30, 3637 CrossRef CAS;
(k) S. Chen, L.-J. Chen, H.-B. Yang, H. Tian and W. Zhu, J. Am. Chem. Soc., 2012, 134, 13596 CrossRef CAS PubMed;
(l) X.-D. Xu, J. Zhang, L.-J. Chen, R. Guo, D.-X. Wang and H.-B. Yang, Chem. Commun., 2012, 48, 11223 RSC;
(m) Q. Han, L.-L. Wang, Q.-J. Li, G.-Z. Zhao, J. He, B. Hu, H. Tan, Z. Abliz, Y. Yu and H.-B. Yang, J. Org. Chem., 2012, 77, 3426 CrossRef CAS PubMed;
(n) L.-J. Chen, Q.-J. Li, J. He, H. Tan, Z. Abliz and H.-B. Yang, J. Org. Chem., 2012, 77, 1148 CrossRef CAS PubMed;
(o) Q.-J. Li, G.-Z. Zhao, L.-J. Chen, H. Tan, C.-H. Wang, D.-X. Wang, D. A. Lehman, D. C. Muddiman and H.-B. Yang, Organometallics, 2012, 31, 7241 CrossRef CAS;
(p) N.-W. Wu, Q.-J. Li, J. Zhang, J. He, J.-K. Ou-Yang, H. Tan, Z. Abliz and H.-B. Yang, Tetrahedron, 2013, 69, 5981 CrossRef CAS PubMed;
(q) X.-D. Xu, J.-K. Ou-Yang, J. Zhang, Y.-Y. Zhang, H.-Y. Gong, Y. Yu and H.-B. Yang, Tetrahedron, 2013, 69, 1086 CrossRef CAS PubMed;
(r) J.-K. Ou-Yang, L.-J. Chen, L. Xu, C.-H. Wang and H.-B. Yang, Chin. Chem. Lett., 2013, 24, 471 CrossRef CAS PubMed.
-
(a) Z. Chu, C. A. Dreissb and Y. Feng, Chem. Soc. Rev., 2013, 42, 7174 RSC;
(b) Y. D. Liu and H. J. Choi, Soft Matter, 2012, 8, 11961 RSC;
(c) J. Hu, G. Zhang and S. Liu, Chem. Soc. Rev., 2012, 41, 5933 RSC.
-
(a) P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133 CrossRef CAS PubMed;
(b) L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201 CrossRef CAS PubMed;
(c) F. Fages, Top. Curr. Chem., 2005, 256, 1 CrossRef;
(d) N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821 RSC;
(e) M. George and R. G. Weiss, Acc. Chem. Res., 2006, 39, 489 CrossRef CAS PubMed;
(f) Z. Yang, G. Liang and B. Xu, Acc. Chem. Res., 2008, 41, 315 CrossRef CAS PubMed;
(g) P. Dastidar, Chem. Soc. Rev., 2008, 37, 2699 RSC;
(h) M. Suzuki and K. Hanabusa, Chem. Soc. Rev., 2009, 38, 967 RSC.
-
(a) A. Ajayaghosh and V. K. Praveen, Acc. Chem. Res., 2007, 40, 644 CrossRef CAS PubMed;
(b) A. Ajayaghosh, V. K. Praveen and C. Vijayakumar, Chem. Soc. Rev., 2008, 37, 109 RSC;
(c) X. Dou, W. Pisula, J. Wu, G. J. Bodwell and K. Müllen, Chem.–Eur. J., 2007, 14, 240 CrossRef PubMed;
(d) H. Maeda, Y. Haketa and T. Nakanishi, J. Am. Chem. Soc., 2007, 129, 13661 CrossRef CAS PubMed;
(e) X. Yang, R. Lu, P. Xue, B. Li, D. Xu, T. Xu and Y. Zhao, Langmuir, 2008, 24, 13730 CrossRef CAS PubMed;
(f) G. Palui and A. Banerjee, J. Phys. Chem. B, 2008, 112, 10107 CrossRef CAS PubMed;
(g) D. D. Díaz, J. J. Cid, P. Vázquez and T. Torres, Chem.–Eur. J., 2008, 14, 9261 CrossRef PubMed;
(h) D.-C. Lee, K. K. McGratha and K. Jang, Chem. Commun., 2008, 3636 RSC;
(i) Y.-T. Shen, C.-H. Li, K.-C. Chang, S.-Y. Chin, H.-A. Lin, Y.-M. Liu, C.-Y. Hung, H.-F. Hsu and S.-S. Sun, Langmuir, 2009, 25, 8714 CrossRef CAS PubMed;
(j) L.-J. Chen, J. Zhang, J. He, X.-D. Xu, N.-W. Wu, D.-X. Wang, Z. Abliz and H.-B. Yang, Organometallics, 2011, 30, 5590 CrossRef CAS;
(k) J. Zhang, X.-D. Xu, L.-J. Chen, Q. Luo, N.-W. Wu, D.-X. Wang, X.-L. Zhao and H.-B. Yang, Organometallics, 2011, 30, 4032 CrossRef CAS;
(l) C. Giansante, G. Raffy, C. Schäfer, H. Rahma, M.-T. Kao, A. G. L. Olive and A. D. Guerzo, J. Am. Chem. Soc., 2011, 133, 316 CrossRef CAS PubMed;
(m) K. K. Kartha, S. S. Babu, S. Srinivasan and A. Ajayaghosh, J. Am. Chem. Soc., 2012, 134, 4834 CrossRef CAS PubMed;
(n) A. Jain, K. V. Rao, C. Kulkarni, A. George and S. J. George, Chem. Commun., 2012, 48, 1467 RSC;
(o) Y. Zhang, B. Zhang, Y. Kuang, Y. Gao, J. Shi, X. X. Zhang and B. Xu, J. Am. Chem. Soc., 2013, 135, 5008 CrossRef CAS PubMed;
(p) Z. Zhao, J. W. Y. Lam and B. Z. Tang, Soft Matter, 2013, 9, 4564 RSC;
(q) B. Jiang, L.-J. Chen, L. Xu, S.-Y. Liu and H.-B. Yang, Chem. Commun., 2013, 49, 6977 RSC;
(r) P. Rajamalli and E. Prasad, Langmuir, 2013, 29, 1609 CrossRef CAS PubMed;
(s) S. Yagai, K. Ishiwatari, X. Lin, T. Karatsu, A. Kitamura and S. Uemura, Chem.–Eur. J., 2013, 19, 6971 CrossRef CAS PubMed.
-
(a) M. Shirakawa, S.-i. Kawano, N. Fujita, K. Sada and S. Shinkai, J. Org. Chem., 2003, 68, 5037 CrossRef CAS PubMed;
(b) H. Jintoku, T. Sagawa, T. Sawada, M. Takafuji, H. Hachisako and H. Ihara, Tetrahedron Lett., 2008, 49, 3987 CrossRef CAS PubMed.
-
(a) Y. Kamikawa and T. Kato, Langmuir, 2007, 23, 274 CrossRef CAS PubMed;
(b) S. Diring, F. Camerel, B. Donnio, T. Dintzer, S. Toffanin, R. Capelli, M. Muccini and R. Ziessel, J. Am. Chem. Soc., 2009, 131, 18177 CrossRef CAS PubMed;
(c) B. Adhikari, J. Nanda and A. Banerjee, Chem.–Eur. J., 2011, 17, 11488 CrossRef CAS PubMed;
(d) J. R. Moffat and D. K. Smith, Chem. Commun., 2011, 47, 11864 RSC;
(e) X.-D. Xu, J. Zhang, X. Yu, L.-J. Chen, D.-X. Wang, T. Yi, F. Li and H.-B. Yang, Chem.–Eur. J., 2012, 18, 16000 CrossRef CAS PubMed;
(f) N. Yan, Z. Xu, K. K. Diehn, S. R. Raghavan, Y. Fang and R. G. Weiss, Langmuir, 2013, 29, 793 CrossRef CAS PubMed;
(g) L. Zhang, C. Liu, Q. Jin, X. Zhu and M. Liu, Soft Matter, 2013, 9, 7966 RSC.
-
(a) F. Würthner, Chem. Commun., 2004, 1564 RSC;
(b) X.-Q. Li, X. Zhang, S. Ghosh and F. Würthner, Chem.–Eur. J., 2008, 14, 8074 CrossRef CAS PubMed;
(c) H. Wu, L. Xue, Y. Shi, Y. Chen and X. Li, Langmuir, 2011, 27, 3074 CrossRef CAS PubMed;
(d) P. K. Sukul, D. Asthana, P. Mukhopadhyay, D. Summa, L. Muccioli, C. Zannoni, D. Beljonne, A. E. Rowan and S. Malik, Chem. Commun., 2011, 47, 11858 RSC.
-
(a) V. K. Praveen, S. J. George, R. Varghese, C. Vijayakumar and A. Ajayaghosh, J. Am. Chem. Soc., 2006, 128, 7542 CrossRef CAS PubMed;
(b) A. Ajayaghosh, C. Vijayakumar, R. Varghese and S. J. George, Angew. Chem., Int. Ed., 2006, 45, 456 CrossRef CAS PubMed;
(c) A. Ajayaghosh, V. K. Praveen, S. Srinivasan and R. Varghese, Adv. Mater., 2007, 19, 411 CrossRef CAS.
-
(a) J. Wu, T. Yi, Q. Xia, Y. Zou, F. Liu, J. Dong, T. Shu, F. Li and C. Huang, Chem.–Eur. J., 2009, 15, 6234 CrossRef CAS PubMed;
(b) X. Cao, J. Zhou, Y. Zou, M. Zhang, X. Yu, S. Zhang, T. Yi and C. Huang, Langmuir, 2011, 27, 5090 CrossRef CAS PubMed.
-
(a) L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201 CrossRef CAS PubMed;
(b) M. O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960 CrossRef CAS PubMed;
(c) L. Qin, P. Duan, F. Xie, L. Zhang and M. Liu, Chem. Commun., 2013, 49, 10823 RSC.
-
(a) J. W. Chung, B.-K. An and S. Y. Park, Chem. Mater., 2008, 20, 6750 CrossRef CAS;
(b) A. Y.-Y. Tam, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2009, 131, 6253 CrossRef CAS PubMed;
(c) O. Simalou, X. Zhao, R. Lu, P. Xue, X. Yang and X. Zhang, Langmuir, 2009, 25, 11255 CrossRef CAS PubMed.
-
(a) J. Eastoe, M. Sánchez-Dominguez, P. Wyatt and R. K. Heenan, Chem. Commun., 2004, 2608 RSC;
(b) T. Suzuki, S. Shinkai and K. Sada, Adv. Mater., 2006, 18, 1043 CrossRef CAS;
(c) N. S. S. Kumar, S. Varghese, G. Narayan and S. Das, Angew. Chem., Int. Ed., 2006, 45, 6317 CrossRef CAS PubMed;
(d) S. Wang, W. Shen, Y. L. Feng and H. Tian, Chem. Commun., 2006, 1497 RSC;
(e) J. H. Kim, M. Seo, Y. J. Kim and S. Y. Kim, Langmuir, 2009, 25, 1761 CrossRef CAS PubMed;
(f) Z. Qiu, H. Yu, J. Li, Y. Wang and Y. Zhang, Chem. Commun., 2009, 3342 RSC;
(g) Q. Chen, Y. Feng, D. Zhang, G. Zhang, Q. Fan, S. Sun and D. Zhu, Adv. Funct. Mater., 2010, 20, 36 CrossRef CAS;
(h) X. Ran, H. Wang, P. Zhang, B. Bai, C. Zhao, Z. Yu and M. Li, Soft Matter, 2011, 7, 8561 RSC;
(i) X.-D. Xu, J. Zhang, L.-J. Chen, X.-L. Zhao, D.-X. Wang and H.-B. Yang, Chem.–Eur. J., 2012, 18, 1659 CrossRef CAS PubMed;
(j) D. K. Maiti and A. Banerjee, Chem.–Asian J., 2013, 8, 113 CrossRef CAS PubMed.
-
(a) T. Naota and H. Koori, J. Am. Chem. Soc., 2005, 127, 9324 CrossRef CAS PubMed;
(b) Y. He, Z. Bian, C. Kang, R. Jin and L. Gao, New J. Chem., 2009, 33, 2073 RSC;
(c) G. Cravotto and P. Cintas, Chem. Soc. Rev., 2009, 38, 2684 RSC;
(d) C. Dou, D. Chen, J. Iqbal, Y. Yuan, H. Zhang and Y. Wang, Langmuir, 2011, 27, 6323 CrossRef CAS PubMed.
-
(a) Y. G. Li, K. G. Liu, J. Liu, J. X. Peng, X. L. Feng and Y. Fang, Langmuir, 2006, 22, 7016 CrossRef CAS PubMed;
(b) N. Shi, H. Dong, G. Yin, Z. Xu and S. Li, Adv. Funct. Mater., 2007, 17, 1837 CrossRef CAS;
(c) D.-C. Lee, K. K. McGrath and K. Jang, Chem. Commun., 2008, 3636 RSC.
-
(a) S.-I. Kawano, N. Fujita and S. Shinkai, J. Am. Chem. Soc., 2004, 126, 8592 CrossRef CAS PubMed;
(b) C. Wang, Q. Chen, F. Sun, D. Zhang, G. Zhang, Y. Huang, R. Zhao and D. Zhu, J. Am. Chem. Soc., 2010, 132, 3092 CrossRef CAS PubMed.
-
(a) J. E. A. Webb, M. J. Crossley, P. Turner and P. Thordarson, J. Am. Chem. Soc., 2007, 129, 7155 CrossRef CAS PubMed;
(b) T. H. Kim, M. S. Choi, B.-H. Sohn, S.-Y. Park, W. S. Lyoo and T. S. Lee, Chem. Commun., 2008, 2364 RSC;
(c) H. Maeda, Chem.–Eur. J., 2008, 14, 11274 CrossRef CAS PubMed;
(d) G. O. Lloyd and J. W. Steed, Nat. Chem., 2009, 1, 437 CrossRef CAS PubMed;
(e) P. Rajamalli and E. Prasad, Org. Lett., 2011, 13, 3714 CrossRef CAS PubMed;
(f) Y. Zhang and S. Jiang, Org. Biomol. Chem., 2012, 10, 6973 RSC;
(g) G.-Z. Zhao, L.-J. Chen, W. Wang, J. Zhang, G. Yang, D.-X. Wang, Y. Yu and H.-B. Yang, Chem.–Eur. J., 2013, 19, 10094 CrossRef CAS PubMed;
(h) L.-B. Xing, B. Yang, X.-J. Wang, J.-J. Wang, B. Chen, Q. Wu, H.-X. Peng, L.-P. Zhang, C.-H. Tung and L.-Z. Wu, Langmuir, 2013, 29, 2843 CrossRef CAS PubMed.
- B. F. Lin, K. A. Megley, N. Viswanathan, D. V. Krogstad, L. B. Drews, M. J. Kade, Y. Qian and M.
V. Tirrell, J. Mater. Chem., 2012, 22, 19447 RSC.
- Y. Zhang, X. Guo, W. Si, L. Jia and X. Qian, Org. Lett., 2008, 10, 473 CrossRef CAS PubMed.
-
(a) K. Cantin, S. Rondeau-Gagné, J. R. Néabo, M. Daigle and J.-F. Morin, Org. Biomol. Chem., 2011, 9, 4440 RSC;
(b) A. Roy, M. Maiti, R. R. Nayak and S. Roy, J. Mater. Chem. B, 2013, 1, 5588 RSC.
-
(a) T. M. Figueira-Duarte and K. Müllen, Chem. Rev., 2011, 111, 7260 CrossRef CAS PubMed;
(b) Z.-Y. Li, L. Xu, C.-H. Wang, X.-L. Zhao and H.-B. Yang, Chem. Commun., 2013, 49, 6194 RSC;
(c) N.-W. Wu, J. Zhang, D. Ciren, Q. Han, L.-J. Chen, L. Xu and H.-B. Yang, Organometallics, 2013, 32, 2536 CrossRef CAS;
(d) L. Xu, L.-J. Chen and H.-B. Yang, Chem. Commun. 10.1039/c3cc47484d;
(e) M. He, Q. Han, J. He, Q. Li, Z. Abliz, H. Tan, L. Xu and H. Yang, Chin. J. Chem., 2013, 31, 663 CrossRef CAS;
(f) N.-W. Wu, J. Zhang, C.-H. Wang, L. Xu and H.-B. Yang, Monatsh. Chem., 2013, 144, 553 CrossRef CAS.
-
(a) G. Köhler and K. Rechthaler, Pure Appl. Chem., 1993, 65, 1647 CrossRef;
(b) B. Dietzek, W. Kiefer, G. Hermann, J. Popp and M. Schmitt, J. Phys. Chem. B, 2006, 110, 4399 CrossRef CAS PubMed.
-
(a) L. Xu, Y. Xu, W. Zhu, B. Zeng, C. Yang, B. Wu and X. Qian, Org. Biomol. Chem., 2011, 9, 8284 RSC;
(b) L. Xu, Y. Xu, W. Zhu, X. Sun, Z. Xu and X. Qian, RSC Adv., 2012, 2, 6323 RSC;
(c) L. Xu, Y. Xu, W. Zhu, Z. Xu, M. Chen and X. Qian, New J. Chem., 2012, 36, 1435 RSC;
(d) L. Xu, M.-L. He, H.-B. Yang and X. Qian, Dalton Trans., 2013, 42, 8218 RSC.
-
(a) P. F. Duan, X. F. Zhu and M. H. Liu, Chem. Commun., 2011, 47, 5569 RSC;
(b) J. F. Miravet, B. Escuder, M. D. Segarra-Maset, M. Tena-Solsona, I. W. Hamley, A. Dehsorkhi and V. Castelletto, Soft Matter, 2013, 9, 3558 RSC;
(c) X. Yan, B. Jiang, T. R. Cook, Y. Zhang, J. Li, Y. Yu, F. Huang, H.-B. Yang and P. J. Stang, J. Am. Chem. Soc., 2013, 135, 16813 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra02373k |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.