Novel stereocontrolled amidoglycosylation of alcohols with acetylated glycals and sulfamate ester†
Teiichi Murakami*,
Yukari Sato,
Kyoko Yoshioka and
Mutsuo Tanaka
Biomedical Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba Central 6, Tsukuba, Ibaraki 305-8566, Japan. E-mail: t-murakami@aist.go.jp; Fax: +81 298 61 6122; Tel: +81 298 61 9394
First published on 25th April 2014
Abstract
A regio- and stereo-controlled, one-pot amidoglycosylation of alcohols has been achieved using O-acetylated glycals, trichloroethoxysulfonamide, and iodosobenzene in the presence of a rhodium(II) catalyst. The reaction would proceed via stereoselective intermolecular aziridination of the glycal.
2-N-Acetamido-2-deoxyglycosides, most commonly of the D-glucose and D-galactose series, are widely distributed in living organisms as glycoconjugates (glycolipids, glycoproteins) and glycosaminoglycans. Aminosugars on cell surfaces are assumed to play an important role as receptor ligands for protein molecules such as enzymes, antibodies, and lectins. Except D-glucosamine, most aminosugars, e.g., D-galactosamine, D-mannosamine, disaccharide lactosamine, are rather expensive as starting materials for the chemical synthesis of glycoconjugates and oligosaccharides.
Glycals (1,2-dehydro-sugar derivatives) have proved to be useful synthetic precursors of 2-amino-2-deoxy-O-glycosides by way of N-functionalisation at C-2 accompanied by addition of alcohols to C-1 (aza-glycosylation), and a variety of methods have been developed for the nitrogen transfer to glycals over the last few decades.1 Among them, azido-nitration reactions developed by Lemieux and co-workers2 have been widely used since the reaction of O-protected glycals with sodium azide and cerium(IV) ammonium nitrate provides 2-azido-2-deoxy-1-O-nitro-glycoses regioselectively. However, the stereoselectivities are dependent on the structure of the glycal substrate; the azidonitrations of acetylated glucal derivatives often proceed non-stereoselectively to give both epimers of the azido group at C-2 (gluco-N and manno-N isomers).2,3
In recent years, transition metal-catalysed inter- and intra-molecular aziridinations of alkenes have been developed by using nitrenes as a nitrogen source.4 The highly reactive nitrene species are generated in situ from several types of precursors, e.g., sulfonyliminoiodinanes,5 sulfonamides6/sufamate esters7/carbamate esters8 with iodine(III) compounds, N-(sulfonyloxy)carbamates with base,9 chloramine-T,10 and azido-compounds.11 When the aziridination reactions are applied to glycal derivatives, the corresponding 1,2-aziridines would be formed. The anomeric C-1 position of N-sulfonyl or N-carbonyl 1,2-aziridino-glycosides would be highly electrophilic to react readily with nucleophiles providing 2-amino-2-deoxy-glycoside derivatives. There have been several reports on such aminoglycosylation reactions via aziridine intermediates.4c Rojas and co-workers reported intramolecular aziridinations of 3-O-azidoformyl-12a and 3-O-carbamoyl-D-allal derivatives12b and subsequent reactions with alcohols to access 2-amino-2-deoxy-β-D-allopyranosides stereoselectively. Liu and co-workers reported stereoselective synthesis of glucosamine derivatives via rhodium-catalysed, substrate-controlled aziridination of 4-O- or 6-O-sulfamoyl-D-glucal derivatives.13 However, these precedents require preparation of the appropriate substrates for intramolecular aziridinations. Stereoselective synthesis of 2-amino-2-deoxy-1-O-glycosides from glycals via intermolecular aziridination14 should be more challenging since it would likely afford a mixture of stereoisomers. Indeed, to our knowledge, this type of glycosylation has been reported only by Descotes' group; addition of photochemically generated N-ethoxycarbonyl-nitrenes to acetylated glycals in methanol gave the methyl 2-amino-glycosides as a mixture of three stereoisomers.15
We report here a regio- and stereo-controlled amidoglycosylation of alcohols via intermolecular aziridination in a one-pot manner using simple O-acetylated glycals (D-glucal, D-galactal, D-lactal), which are readily prepared in 3 steps from the parent sugars. In their research on rhodium-catalysed olefin aziridination with PhI(OAc)2 and sulfamate esters, Du Bois and co-workers found a stereospecific amido-acetoxylation of tri-O-acetyl-D-glucal 1a to give 2-deoxy-2-(trichloroethoxysulfonyl)amino-glucopyranosyl 1β-O-acetate 2a-β in a regiospecific manner in high yield,16 though they have not described the reaction in detail. We were interested in the amido-acetoxylation, and confirmed that the reaction of 1a with Cl3CCH2OSO2NH2, PhI(OAc)2, MgO in the presence of a Rh(II) catalyst [Rh2(NHCOCF3)4] afforded the β-acetate 2a-β only. The amidoacetoxylation was applied to acetyl-protected galactal 1b and lactal 1c under identical conditions. As shown in Table 1, Ac-galactal 1b gave a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the α- and β-acetates, whereas Ac-lactal 1c gave the β-acetate 2c-β predominantly. In all cases, the stereoisomers at C-2 were not detected.
:1 mixture of the α- and β-acetates, whereas Ac-lactal 1c gave the β-acetate 2c-β predominantly. In all cases, the stereoisomers at C-2 were not detected.
|
||
|---|---|---|
| 1 | Yield (%) | |
| 2-β | 2-α | |
| a Reagents and conditions: Cl3CCH2OSO2NH2 (1.8 equiv.), PhI(OAc)2 (1.8 equiv.), Rh2(NHCOCF3)4 (0.1 equiv.), MgO (4 equiv.) in PhCl, 5 °C, 2 h to rt, 10 h.b Isolated yield by silica gel chromatography.c The yields were determined by 1H-NMR integration of the α/β mixture. | ||
| a: R1 = AcO, R2 = H | 91b | 0 |
| b: R1 = H, R2 = AcO | 33b | 61b |
 |
84c | 8c |
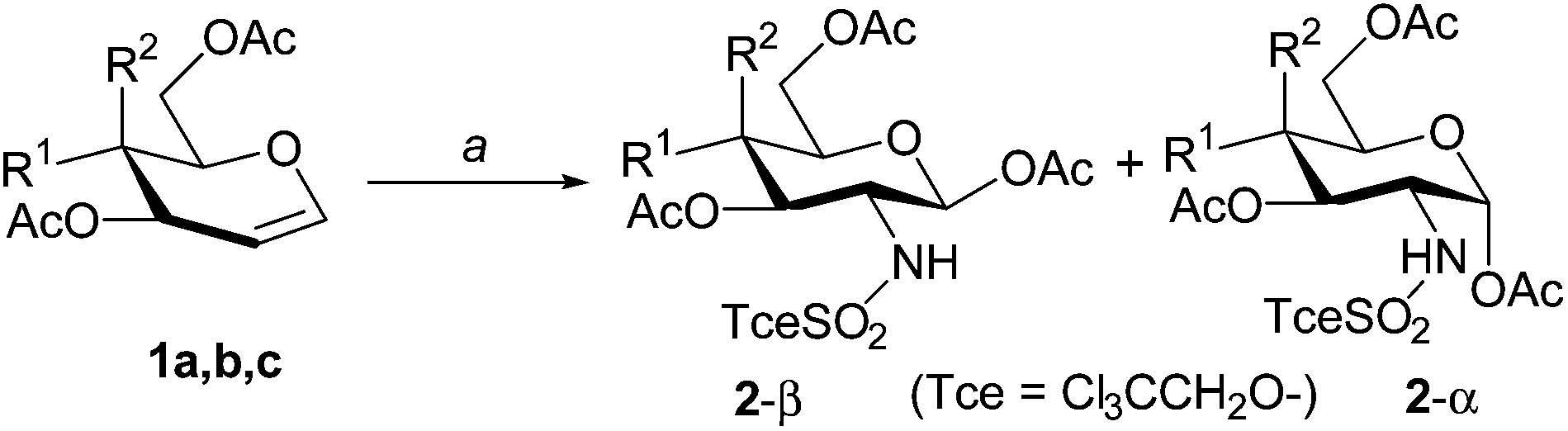
We next examined a direct synthesis of 2-sulfonamido-1-O-glycosides17 from glycals by adding alcohols in the reaction mixture. As shown in Table 2, the reaction of 1a and tetradecanol (4 equiv.)12 with Cl3CCH2OSO2NH2 in the presence of PhI(OAc)2 and catalytic Rh2(NHCOCF3)4 in chlorobenzene afforded the desired tetradecyl-β-glucoside 3a in 58% yield with no α-glucoside. However, the 1-O-acetate 2a-β was also formed (entry 1). Formation of 2a was suppressed by using iodosobenzene with 4 Å molecular sieves (as dehydration agent) in place of PhI(OAc)2 with MgO (entry 2). Reduction of the amount of alcohol improved the yield of 3a (entry 3). For the aziridination catalyst, Rh2(OAc)4 was less effective than Rh2(NHCOCF3)4, and copper(I)6a and silver18 catalysts were much less effective (entries 4–6).
|
||||
|---|---|---|---|---|
| Entry | Equiv. of C14H29OH | Oxidanta | Catalyst | 3a Yieldb (%) |
| a The oxidant (solid, 1.8 equiv.) was added in ca. 6 portions to the mixture of other reactants for ca. 1 h at 5 °C.b Isolated yield by silica gel chromatography. In all cases, unreacted 1a remained.c MgO (4 equiv.) was used in place of MS4A.d 1-Acetate 2a-β was obtained in 22% yield.e tBu3tpy: 4,4′,4′′-tri-tert-butyl-2,2′:6′,2′′-terpyridine. | ||||
| 1c | 4 | PhI(OAc)2 | Rh2(NHCOCF3)4 | 58d |
| 2 | 4 | PhI![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
Rh2(NHCOCF3)4 | 66 |
| 3 | 2 | PhIO |
Rh2(NHCOCF3)4 | 78 |
| 4 | 4 | PhIO |
Rh2(OAc)4 | 45 |
| 5 | 2 | PhIO |
Cu(CH3CN)4PF6 | 17 |
| 6 | 2 | PhIO |
AgNO3, tBu3tpye | 25 |

With the optimised reaction conditions in hand, we investigated the scope and generality of this Rh-catalysed one-pot amidoglycosylation, and the results are summarized in Table 3. Reactions of 12-bromododecanol and 2-phenylethanol with the glycals 1a–c proceeded smoothly to afford the corresponding β-glycosides 4a, b, c, 6a in good yields (entries 1, 2, 3 and 5). In contrast, 12-acetylthio-1-dodecanol gave the galactoside 5b in poor yield under identical conditions, indicating that the acetylthio group would suppress the reaction (entry 4). Reaction of 1b with 4-penten-1-ol gave the β-galactoside 8b in somewhat lower yield along with a byproduct‡ derived from 4-pentenol, indicating that pentenol was also aziridinated, but would be less reactive than 1b (entry 7).
|
||||
|---|---|---|---|---|
| Entry | Glycal | R–OH | Product | Yieldb (%) |
| a General procedure: to a mixture of glycal 1, ROH (2 equiv.), Cl3CCH2OSO2NH2 (1.7 equiv.), Rh2(NHCOCF3)4 (0.1 equiv.), molecular sieves 4 Å (0.8 g mmol−1) in PhCl (1: 0.05–0.10 M) under nitrogen at 5 °C was added PhIO (1.8 equiv.) in several portions for 1 h, and the resulting suspension was stirred at 5 °C for 1 h and then at rt for 5–15 h.b Isolated yield by silica gel chromatography. | ||||
| 1 | 1a | Br(CH2)12–OH | 4a | 77 |
| 2 | 1b | Br(CH2)12–OH | 4b | 84 |
| 3 | 1c | Br(CH2)12–OH | 4c | 70 |
| 4 | 1b | AcS(CH2)12–OH | 5b | 11 |
| 5 | 1a | Ph(CH2)2–OH | 6a | 75 |
| 6 | 1b | PhCH2–OH | 7b | 62 |
| 7 | 1b | H2CCH–(CH2)3–OH |
8b | 63 |
| 8 | 1a | Cyclohexanol | 9a | 57 |
| 9 | 1b | Cyclohexanol | 9b | 78 |
| 10 | 1a |  |
10a | 74 |
| 11 | 1b | L(−)-Menthol | 10b | 76 |
| 12 | 1c | L(−)-Menthol | 10c | 67 |
| 13 | 1b |  |
11b | 74 |
| 14 | 1b |  |
12b | 21 |
| 15 | 1b |  |
13b | 56 |

Cyclic secondary alcohols: cyclohexanol and L(−)-menthol reacted with the glycals 1a–c to give the corresponding β-glycosides in good yields. Ac-galactal 1b appeared to be more reactive and gave the glycosides in better yields than 1a and 1c (entries 8–12 and 1–3). For sugar-derived alcohols, 1,2:3,4-di-O-isopropylidene-α-D-galactopyranose afforded the galactoside 11b in good yield, whereas methyl 2,3,4-tri-O-benzyl-α-D-glucopyranoside gave the galactoside 12b in low yield, and substantial amounts of some byproducts: de-O-benzylated glucoses and benzaldehyde were obtained. In all the cases examined, no α-anomer was detected by 1H- and 13C-NMR. Small amounts (3–8%) of hydrolysed products (1-OH) were formed in most cases.
The reaction would proceed via the generation of sulfonyl-nitrene followed by formation of the α-oriented aziridine intermediate (14) presumably due to the presence of β-acetoxy group at C-3.19 The aziridine would be opened with the alcohol at C-1 from the β-face to afford 1,2- and 2,3-di-trans-product (Scheme 1).
 | ||
| Scheme 1 Proposed reaction pathway. | ||
The trichloroethoxysulfonyl group in 3a was removed by treatment with zinc and acetic acid in the presence of CuSO4 to give the free amine 15. When the desulfonylation was carried out in the presence of acetic anhydride, the acetamide 16 was obtained in good yield (Scheme 2).
 | ||
| Scheme 2 Deprotection of the trichloroethoxysulfonyl group. | ||
In conclusion, we have developed a regio- and stereo-selective synthesis of 2-amino-2-deoxy-1-O-β-glycosides from acetylated glycals via rhodium(II)-catalysed intermolecular aziridination with trichloroethoxysulfonamide and iodosobenzene. This amidoglycosylation proceeds smoothly under mild conditions without the use of usual O-glycosylation promoters such as Lewis acids, and is applicable to a variety of primary and secondary alcohols.
Notes and references
- For a review, see: A. F. G. Bongat and A. V. Demchenko, Carbohydr. Res., 2007, 342, 374 CrossRef CAS PubMed.
- (a) R. U. Lemieux and R. M. Ratcliffe, Can. J. Chem., 1979, 57, 1244 CrossRef CAS PubMed; (b) R. U. Lemieux, S. Z. Abbas, M. H. Burzynska and R. M. Ratcliffe, Can. J. Chem., 1981, 60, 63 CrossRef.
- (a) C.-H. Lin, T. Sugai, R. L. Halcomb, Y. Ichikawa and C.-H. Wong, J. Am. Chem. Soc., 1992, 114, 10138 CrossRef CAS; (b) P. H. Seeberger, S. Roehrig, P. Schell, Y. Wang and W. J. Christ, Carbohydr. Res., 2000, 328, 61 CrossRef CAS; (c) T. Murakami, K. Yoshioka, Y. Sato, M. Tanaka, O. Niwa and S. Yabuki, Bioorg. Med. Chem. Lett., 2011, 21, 1265 CrossRef CAS PubMed.
- For reviews, see: (a) P. Müller and C. Fruit, Chem. Rev., 2003, 103, 2905 CrossRef PubMed; (b) H. Pellissier, Tetrahedron, 2010, 66, 1509 CrossRef CAS PubMed; (c) G. Dequirez, V. Pons and P. Dauban, Angew. Chem., Int. Ed., 2012, 51, 7384 CrossRef CAS PubMed.
- For a review, see: P. Dauban and R. H. Dodd, Synlett, 2003, 1571 CrossRef CAS PubMed.
- (a) P. Dauban, L. Sanière, A. Tarrade and R. H. Dodd, J. Am. Chem. Soc., 2001, 123, 7707 CrossRef CAS; (b) H.-L. Kwong, D. Liu, K.-Y. Chan, C.-S. Lee, K.-H. Huang and C.-M. Che, Tetrahedron Lett., 2004, 45, 3965 CrossRef CAS PubMed; (c) R. Fan, D. Pu, J. Gan and B. Wang, Tetrahedron Lett., 2008, 49, 4925 CrossRef CAS PubMed.
- (a) K. Guthikonda and J. Du Bois, J. Am. Chem. Soc., 2002, 124, 13672 CrossRef CAS PubMed; (b) Q. Xu and D. H. Appella, Org. Lett., 2008, 10, 1497 CrossRef CAS PubMed; (c) S. Beaumont, V. Pons, P. Rtailleau, R. H. Dodd and P. Dauban, Angew. Chem., Int. Ed., 2010, 49, 1634 CrossRef CAS PubMed.
- (a) A. Padwa and T. Stengel, Org. Lett., 2002, 4, 2137 CrossRef CAS PubMed; (b) C. J. Hayes, P. W. Beavisa and L. A. Humphries, Chem. Commun., 2006, 4501 RSC.
- (a) M. Carducci, S. Fioravanti, M. A. Loreto, L. Pellacani and P. A. Tardella, Tetrahedron Lett., 1996, 37, 3777 CrossRef CAS; (b) H. Lebel, S. Lectard and M. Parmentier, Org. Lett., 2007, 9, 4797 CrossRef CAS PubMed.
- (a) T. Ando, S. Minakata, I. Ryu and M. Komatsu, Tetrahedron Lett., 1998, 39, 309 CrossRef CAS; (b) J. U. Jeong, B. Tao, I. Sagasser, H. Henniges and K. B. Sharpless, J. Am. Chem. Soc., 1998, 120, 6844 CrossRef CAS; (c) S. Minakata, D. Kano, Y. Oderaotoshi and M. Komatsu, Angew. Chem., Int. Ed., 2004, 43, 79 CrossRef PubMed.
- (a) S. C. Bergmeier and D. M. Stanchina, J. Org. Chem., 1997, 62, 4449 CrossRef CAS PubMed; (b) K. Omura, T. Uchida, R. Irie and T. Katsuki, Chem. Commun., 2004, 2060 RSC; (c) G.-Y. Gao, J. E. Jones, R. Vyas, J. D. Harden and X. P. Zhang, J. Org. Chem., 2005, 71, 6655 CrossRef PubMed.
- (a) C. Kan, C. M. Long, M. Paul, C. M. Ring, S. E. Tully and C. M. Rojas, Org. Lett., 2001, 3, 381 CrossRef CAS; (b) E. Levites-Agababa, E. Menhaji, L. N. Perison and C. M. Rojas, Org. Lett., 2002, 4, 863 CrossRef CAS PubMed.
- (a) R. Lorpitthaya, Z.-Z. Xie, J.-L. Kuo and X.-W. Liu, Chem.–Eur. J., 2008, 14, 1561 CrossRef CAS PubMed; (b) R. Lorpitthaya, K. B. Sophy, J.-L. Kuo and X.-W. Liu, Org. Biomol. Chem., 2009, 7, 1284 RSC.
- Stereoselective synthesis of 1,2-trans-1,2-bis(tosylamino)-glycoside derivatives (N-glycosides) from glycals via intermolecular aziridination with chloramine-T was reported; V. Kumar and N. G. Ramesh, Chem. Commun., 2006, 4952 RSC.
- (a) E. Kozlowska-Gramsz and G. Descotes, Tetrahedron Lett., 1981, 22, 563 CrossRef CAS; (b) E. Kozlowska-Gramsz and G. Descotes, Can. J. Chem., 1982, 60, 558 CrossRef CAS.
- K. Guthikonda, P. M. Wehn, B. J. Caliando and J. Du Bois, Tetrahedron, 2006, 62, 11331 CrossRef CAS PubMed.
- A two-step sulfonamidoglycosylation from glycals was reported; D. A. Griffith and S. J. Danishefsky, J. Am. Chem. Soc., 1990, 112, 5811 CrossRef CAS.
- Y. Cui and C. He, J. Am. Chem. Soc., 2003, 125, 16202 CrossRef CAS PubMed.
- For related 2,3-trans-selective additions to glycals, see: (a) J. Du Bois, C. S. Tomooka, J. Hong and E. M. Carreira, J. Am. Chem. Soc., 1997, 119, 3179 CrossRef CAS; (b) J. Liu and D. Y. Gin, J. Am. Chem. Soc., 2002, 124, 9789 CrossRef CAS PubMed; (c) R. S. Dahl and N. S. Finney, J. Am. Chem. Soc., 2004, 126, 8356 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data, copies of 1H and 13C NMR, and mass spectra. See DOI: 10.1039/c4ra02367f |
| ‡ 2-(Trichloroethoxysulfonylamino)methyltetrahydrofuran was obtained in ca. 0.5 molar ratio to major product 8b. |
| This journal is © The Royal Society of Chemistry 2014 |