A non-covalent complex based on catechol–benzoxazole moieties: electrochemical synthesis and characterization†
Hamid Salehzadeh and
Davood Nematollahi*
Faculty of Chemistry, Bu-Ali Sina University, Hamedan, 65178-38683, Iran. E-mail: nemat@basu.ac.ir; Fax: +98-811-8257407
First published on 23rd May 2014
Abstract
Electrochemical oxidation of 3,5-di-tert-butylcatechol has been studied in the presence of 2-methoxybenzylamine by means of cyclic voltammetry and controlled-potential coulometry. The results indicate that the electrogenerated 3,5-di-tert-butylcyclohexa-3,5-diene-1,2-dione participates in a cyclization reaction with 2-methoxybenzylamine and converts to the corresponding benzoxazole (4). Afterward by a non-covalent interaction, a proton transfer complex is formed between benzoxazole 4 and 3,5-di-tert-butylcatechol.
Introduction
As heteroaromatic systems, benzoxazole derivatives are important structural motifs in many biological, pharmaceutical and dye compounds.1 They are used as cytotoxic agents,2 cathepsin S inhibitors,3 HIV reverse transcriptase inhibitors,4 estrogen receptor agonists,5 selective peroxisome proliferator activated receptor antagonists,6 anticancer agents,7 and orexin-1 receptor antagonists.8In this connection, we have recently reported a new strategy for the electrochemical synthesis of some 5,7-di-tert-butyl-2-phenylbenzo[d]oxazole derivatives based on the reaction of benzylamine derivatives with 3,5-di-tert-butylcyclohexa-3,5-diene-1,2-dione.9 In the present work, we have studied the electrochemical oxidation of 3,5-di-tert-butylcatechol in the presence 2-methoxybenzylamine to represent an electrochemical synthesis of a new non-covalent complex containing 5,7-di-tert-butyl-2-(2-methoxyphenyl) benzo[d] oxazole and 3,5-di-tert-butylcatechol.
Results and discussion
Electrochemical oxidation of 3,5-di-tert-butylcatechol (DTC) in the absence and presence of 2-methoxybenzylamine (MBA) as a nucleophile was studied in some details. The cyclic voltammogram of DTC in aqueous phosphate buffer (0.2 M, pH 7.5)–ethanol mixture (80/20 v/v) shows one anodic peak (A1) and its counterpart cathodic peak (C1) which correspond to the transformation of DTC to 3,5-di-tert-butylcyclohexa-3,5-diene-1,2-dione (DBQ) and vice versa within a quasi-reversible two-electron process (Fig. 1, curve a).9,10 Curve b in Fig. 1 shows the cyclic voltammogram of DTC in the presence of MBA. The occurrence of a chemical reaction after the electrochemical oxidation is supported by decrease in the current of peak C1 during the reverse scan, which could indicate that DBQ formed at the surface of the electrode is consumed by a chemical reaction with MBA.9 More studies were performed by varying the potential sweep rate of cyclic voltammograms of DTC in the presence of MBA. The results indicate that the peak current ratio (IpC1/IpA1) increases by increasing sweep rate. With increasing potential scan rate, the time required for the reaction of DBQ with MBA is not enough, and consequently, the peak current ratio (IpC1/IpA1) increases.9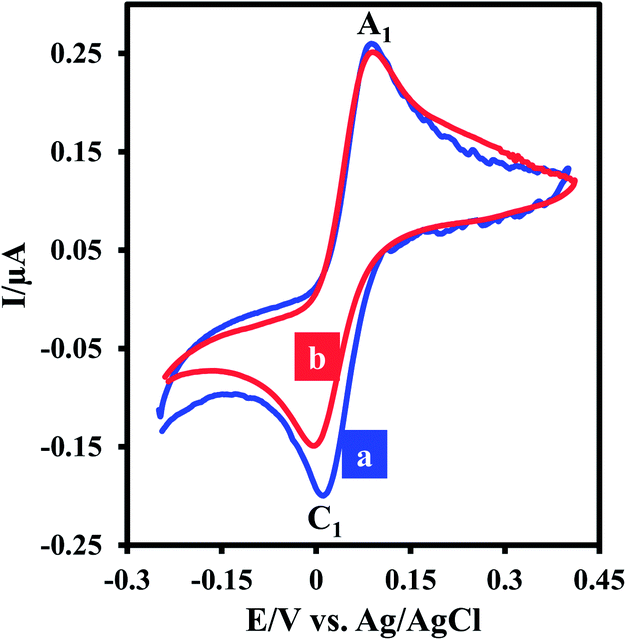 | ||
| Fig. 1 Cyclic voltammograms of DTC (0.1 mM): (a) in the absence and (b) in the presence of MBA (30 mM) at a glassy carbon electrode, in aqueous phosphate buffer (0.2 M, pH 7.5)–ethanol mixture (80/20 v/v). Scan rate: 2.5 mV s−1; t = 25 ± 1 °C. | ||
Controlled-potential coulometry was performed in aqueous phosphate buffer (c = 0.1 M. pH = 7.5)–ethanol (v/v 80/20) mixture containing DTC and MBA at 0.15 V versus Ag/AgCl. Fig. 2 presents the cyclic voltammograms during controlled potential coulometry. The plot of IpA1 vs. Q is shown in the inset of Fig. 2. As can be seen, IpA1 and IpC1 decrease during coulometry and both disappear when the charge consumption becomes 2e− per molecule of DTC.
 | ||
| Fig. 2 Cyclic voltammograms of DTC (0.25 mmol) in the presence of MBA (0.125 mmol) during controlled potential coulometry at 0.15 V versus Ag/AgCl in aqueous phosphate buffer (0.2 M, pH 7.5)–ethanol mixture (80/20 v/v), after consumption of: (a) 0, (b) 10, (c) 20, (d) 30 and (e) 40 C. Scan rate: 100 mV s−1. Inset: variation of IpA1 versus charge consumed during controlled potential coulometry. | ||
Diagnostic criteria of cyclic voltammetry and controlled potential coulometry accompanied by 1H NMR, 13C NMR, MS (see ESI†) and X-ray spectra of final product (Fig. 3) allow us to propose the following mechanism for the electrochemical oxidation of DTC in the presence of MBA (Scheme 1). According to our results, the condensation reaction of MBA with electrogenerated DBQ at the most electrophilic carbonyl, leads to intermediate 1 (imine formation). Rearrangement then led to the formation of the Schiff base 2. In the next step, via a cyclization reaction, 2 is converted to benzoxazoline 3. Chemical or electrochemical oxidative dehydrogenation of 3 in the next step leads to the corresponding benzoxazole 4. The affinity of the non-covalent interaction between 4 and DTC causes the formation of non-covalent complex 5.
 | ||
| Fig. 3 Single-crystal X-ray diffraction of 5. | ||
 | ||
| Scheme 1 Proposed mechanism for the electrochemical oxidation of DTC in the presence of MBA. | ||
Non-covalent complexes are highly useful for the development of new molecular electronic devices and optical sensors.11 Quinhydrone is a well-known stable non-covalent complex formed between p-benzoquinone and 1,4-hydroquinone.12 In this complex, the charge transfer and hydrogen-bonding interactions between the electron donor (hydroquinone) and the electron acceptor (quinone) stabilize the complex. In addition, another quinhydrone complex was described by the reaction of phenanthrene-9,10-dione with phenanthrene-9,10-diol (ortho-quinhydrone).13 We also reported a non-covalent complex based on 3,5-di-tert-butylcatechol and its oxidized form, 3,5-di-tert-butylcyclohexa-3,5-diene-1,2-dione.10
Fig. 4 compares the cyclic voltammograms of DTC and non-covalent complex 5. As can be seen, the oxidation potential of 3,5-di-tert-butylcatechol moiety of 5 is more positive than that of DTC (EpA2 > EpA1). In contrast, in the reverse scan, the potential of the two cathodic peaks (C1) are nearly same. This is expected because the hydrogen bonding makes 5 difficult to oxidation. On the other hand, since there is no interaction between DBQ and benzoxazole 4, both cathodic peaks have same potential.
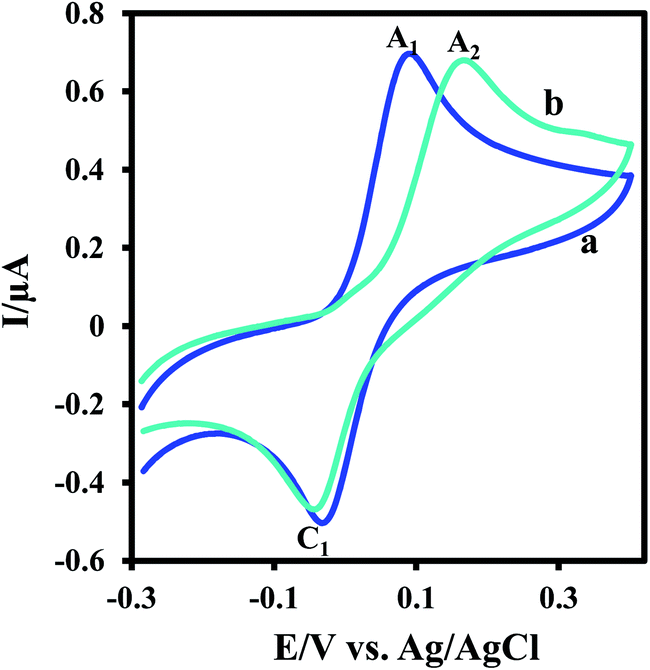 | ||
| Fig. 4 Cyclic voltammogram of (a) DTC (0.5 mM) and (b) non-covalent complex 5 (0.5 mM) at a glassy carbon electrode in aqueous phosphate buffer (0.2 M, pH 7.5)–ethanol mixture (50/50 v/v). Scan rate: 10 mV s−1; t = 25 °C. | ||
The effect of hydrogen bonding between DTC and benzoxazole moieties in 5 has been shown in its FT-IR spectrum. Analysis of the IR spectrum reveals two peaks at 3499 and 3188 cm−1 that are related to two OH groups of bonded DTC to benzoxazole 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 (see ESI†).
10 (see ESI†).
Conclusion
This is the first report showing the formation of a complex between benzoxazole and catechol moieties via an intermolecular hydrogen bonding. Recently, we have shown that DBQ reacts with benzylamine derivatives to form 2-arylbenzoxazoles derivatives.9 However, the results of this work revealed that electro-oxidation of DTC in the presence of MBA gives non-covalent complex 5 whose structure was established by X-ray analysis.Experimental
Cyclic voltammetry, controlled-potential coulometry and preparative electrolysis were performed using an Autolab model PGSTAT 30 potentiostat/galvanostat. The working electrode used in the voltammetry experiments was a glassy carbon disc (1.8 mm2 area) and platinum wire was used as counter electrode. The working electrode used in controlled-potential coulometry and synthesis was an assembly of four carbon rods (31 cm2) and large platinum gauze constitute the counter electrode. The working electrode potentials were measured versus Ag/AgCl/3 M KCl (all electrodes from AZAR electrode). 3,5-Di-tert-butylcatechol, 2-methoxybenzylamine and other solvents and reagents were reagent-grade materials from Aldrich. The glassy carbon electrode was polished using alumina slurry (from Iran Alumina Co.).General procedure for synthesis of 5
For synthesis of 5, a solution (80 mL) of water (phosphate buffer, pH = 7.5, c = 0.2 M)–ethanol (80/20, v/v) containing DTC (1.0 mmol) and MBA (0.5 mmol) was electrolyzed at 0.15 V versus Ag/AgCl, in a divided cell. The electrolysis was terminated when the decay of the current became more than 95% of the initial current. At the end of electrolysis, the precipitated solid was collected by filtration and recrystallized in n-hexane (71% yield). After recrystallization, products were characterized by: MS, IR and 1H NMR, 13C NMR and single-crystal X-ray diffraction (see ESI†). mp = 133–135 °C. IR (KBr, cm−1): 3500, 2962, 2905, 2868, 1603, 1584, 1551, 1481, 1465, 1437, 1362, 1311, 1248, 1122, 1076, 1044, 1022, 968, 870, 855, 752, 655 and 496; 1H NMR (400 MHz, CDCl3) δ 1.21 (s, 9H tert-butyl), 1.38 (s, 9H tert-butyl), 1.46 (s, 9H tert-butyl), 1.58 (s, 9H tert-butyl), 4.00 (s, 3H CH3), 5.98 (s, 1H OH), 6.80 (d, J = 2.0 Hz, 1H Ar), 6.88 (d, J = 2.0 Hz, 1H Ar), 7.10–7.17 (m, 2H Ar), 7.34 (d, J = 2.0 Hz, 1H Ar), 7.52–7.56 (m, 1H Ar), 7.61 (d, J = 1.6 Hz, 1H Ar), 8.17 (dd, Jd = 7.8, Jdd = 1.8 Hz, 1H Ar); 13C NMR (100 MHz, CDCl3) δ 160.9, 158.5, 147.7, 146.3, 143.0, 141.7, 141.5, 141.2, 135.3, 133.6, 132.8, 130.9, 120.7, 119.6, 116.0, 115.3, 114.1, 112.1, 110.6, 55.9, 35.1, 34.8, 34.4, 34.2, 31.8, 31.5, 29.9, 29.7; MS (m/z) (relative intensity): 337.3 (22), 322.3 (24), 222.2 (35), 207.2 (100), 179.1 (6), 82 (6).Acknowledgements
We acknowledge the Bu-Ali Sina University Research Council and Center of Excellence in Development of Environmentally Friendly Methods for Chemical Synthesis (CEDEFMCS) for their support of this work.References
- (a) M. S. Malamas, E. S. Manas, R. E. McDevitt, I. Gunawan, Z. B. Xu, M. D. Collini, C. P. Miller, T. Dinh, R. A. Henderson, J. C. Keith Jr and H. A. Harris, J. Med. Chem., 2004, 47, 5021 CrossRef CAS PubMed; (b) H. Razavi, S. K. Palaninathan, E. T. Powers, R. L. Wiseman, H. E. Purkey, N. N. Mohamedmohaideen, S. Deechongkit, K. P. Chiang, M. T. A. Dendle, J. C. Sacchettini and J. W. Kelly, Angew. Chem., Int. Ed., 2003, 42, 2758 CrossRef CAS PubMed; (c) M. Taki, J. L. Wolford and T. V. O'Halloran, J. Am. Chem. Soc., 2004, 126, 712 CrossRef CAS PubMed; (d) S. Ueda and H. Nagasawa, Angew. Chem., Int. Ed., 2008, 47, 6411 CrossRef CAS PubMed; (e) I. H. Leaver and B. Milligam, Dyes Pigm., 1984, 5, 109 CrossRef CAS.
- (a) J. P. Davidson and E. J. Corey, J. Am. Chem. Soc., 2003, 125, 13486 CrossRef CAS PubMed; (b) S. Sato, T. Kajiura, M. Noguchi, K. Takehana, T. Kobavashi and T. Tsuji, J. Antibiot., 2001, 54, 102 CrossRef CAS.
- D. C. Tully, H. Liu, P. B. Alper, A. K. Chatterjee, R. Epple, M. J. Roberts, J. A. Williams, K. T. Nguyen, D. H. Woodmansee, C. Tumanut, J. Li, G. Spraggon, J. Chang, T. Tuntland, J. L. Harris and D. S. Karanewsky, Bioorg. Med. Chem. Lett., 2006, 16, 1975 CrossRef CAS PubMed.
- J. A. Grobler, G. Dornadula, R. M. Rice, A. L. Simcoe, D. J. Hazuda and M. D. Miller, J. Biol. Chem., 2007, 282, 8005 CrossRef CAS PubMed.
- L. Leventhal, M. R. Brandt, T. A. Cummonts, M. J. Piesla, K. E. Rogers and H. A. Harris, Eur. J. Pharmacol., 2006, 553, 146 CrossRef CAS PubMed.
- J. Nishiu, M. Ito, Y. Ishida, M. Kakutani, T. Shibata, M. Matsushita and M. Shindo, Diabetes, Obes. Metab., 2006, 8, 508 CrossRef CAS PubMed.
- J. Easmon, G. Pürstinger, K. S. Thies, G. Heinisch and J. Hofmann, J. Med. Chem., 2006, 49, 6343 CrossRef CAS PubMed.
- K. Rasmussen, M. A. Hsu and Y. Yang, Neuropsychopharmacology, 2007, 32, 786 CrossRef CAS PubMed.
- H. Salehzadeh, D. Nematollahi and H. Hesari, Green Chem., 2013, 15, 2441 RSC.
- D. Nematollahi and H. Shayani-Jam, Electrochim. Acta, 2006, 51, 6384 CrossRef CAS PubMed.
- (a) D. L. Feldheim and C. D. Keating, Chem. Soc. Rev., 1998, 27, 1 RSC; (b) M. C. Petty, M. R. Bryce and D. Bloor, Introduction of Molecular Electronics, Oxford University Press, New York, 1995 Search PubMed; (c) M. Emmelius, G. Pawlowski and H. W. Vollmann, Angew. Chem., Int. Ed. Engl., 1989, 28, 1445 CrossRef; (d) J. R. Lakowica, Principles of Fluorescence Spectroscopy, Kluwer/Plenum, New York, 2nd edn, 1999 Search PubMed; (e) A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515 CrossRef CAS PubMed.
- (a) B. R. Eggins and J. Q. Chambers, J. Electrochem. Soc., 1970, 117, 186 CrossRef CAS PubMed; (b) R. E. Moser and H. G. Cassidy, J. Am. Chem. Soc., 1965, 87, 3463 CrossRef CAS; (c) V. D. Parker, Chem. Commun., 1969, 716 RSC; (d) F. Foster, Organic Charge–Transfer Complexes, Academic Press, New York, 1969 Search PubMed; (e) M. A. Slifkin, Charge–Transfer Interactions of Biomolecules, Academic Press, New York, 1971 Search PubMed; (f) F. Gutmann, C. Johnson, H. Keyzer and J. Molnar, Charge–Transfer Complexes in Biological Systems, Marcel Dekker, New York, 1997 Search PubMed.
- F. Calderazzo, C. Forte, F. Marchetti, G. Pampaloni and L. Pieretti, Helv. Chim. Acta, 2004, 87, 781 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FT-IR, 1H NMR, 13C NMR, MS spectra and X-ray data of compound 5. CCDC 992193. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra02340d |
| This journal is © The Royal Society of Chemistry 2014 |