
Anhydrous proton motion study by solid state NMR spectroscopy in novel PEMFC blend membranes composed of fluorinated copolymer bearing 1,2,4-triazole functional groups and sPEEK†
Abstract
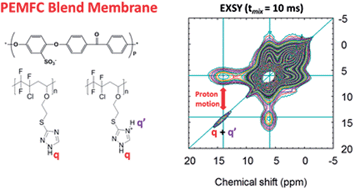
The proton mobility in a new family of PEMFC blend membranes containing 1,2,4-triazole groups is studied by infrared spectroscopy and particularly by 1H Magic Angle Spinning Solid State NMR spectroscopy. These membranes are usually used for fuel cell operation at low relative humidity (RH < 25%). The studied membrane contains 40%-wt of a partially fluorinated alternating poly(2-iodoethyl vinyl ether-alt-chlorotrifluoroethylene)-g-1H-1,2,4-triazole-3-thiol95% copolymer (II) and 60%-wt of sulfonated poly(ether ether ketone), sulfonated PEEK (IEC = 1.3 meq g−1) (r = n–NH/n–SO3H = 1.7). In this study, the 1D 1H MAS spectrum of copolymer (II) was fully determined. Following acidification of a suspension of this copolymer leading to acidified (II′) copolymer, the 1D 1H MAS spectrum showed two populations corresponding to triazole and triazolium groups. The 2D 1H EXchange SpectroscopY spectrum of (II′) showed faster proton dynamics of the triazolium protonated form than in the copolymer containing only non protonated triazole groups. Inhomogeneity of (II′) sample was confirmed by the different spin dynamics of triazole and triazolium ring protons. In the homogeneous membrane, spin dynamics were further increased (i.e. very short mixing time to observe proton diffusion). Hence, this work provides confirmation that protonation acts in favour of proton mobility in the material at room temperature, and provides experimental evidence for the increase of proton mobility due to triazole protonation from the sulfonic acid groups of sPEEK.
 Please wait while we load your content...
Please wait while we load your content...

