
LiV3O8 nanorods as cathode materials for high-power and long-life rechargeable lithium-ion batteries†
Abstract
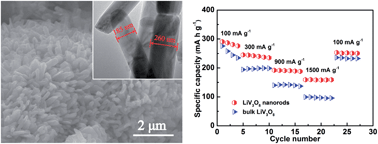
Nowadays one of the principal challenges for the development of lithium-ion batteries (LIBs) is fulfilling the burgeoning demands for high energy and power density with long cycle life. Herein, we demonstrate a two-step route for synthesizing LiV3O8 nanorods with a confined preferential orientation by using VO2(B) nanosheets made in the laboratory as the precursor. The special structures of nanorods endow the LiV3O8 materials with markedly enhanced reversible capacities, high-rate capability and long-term cycling stability as cathodes for lithium storage. The results show that very desirable initial capacities of 161 and 158 mA h g−1 can be achieved for the LiV3O8 nanorods at extremely high rates of 2000 and 3000 mA g−1, with minimal capacity loss of 0.037% and 0.031% per cycle throughout 300 and 500 cycles, respectively. The energetically optimized electron conduction and lithium diffusion kinetics in the electrode process may shed light on the superior electrochemical properties of the LiV3O8 nanorods, primarily benefitting from the small particle size, large surface area and restricted preferential ordering along the (100) plane.
 Please wait while we load your content...
Please wait while we load your content...

