A review of new methods of surface chemical modification, dispersion and electrophoretic deposition of metal oxide particles
M. S. Ata,
Y. Liu and
I. Zhitomirsky*
Department of Materials Science and Engineering, McMaster University, 1280 Main Street West, Hamilton, Ontario L8S 4L7, Canada. E-mail: zhitom@mcmaster.ca; Tel: +1-(905)525-9140
First published on 14th May 2014
Abstract
A bio-inspired chemical approach has been developed for the surface modification, dispersion and electrophoretic deposition (EPD) of metal oxide particles. The study of the chemical mechanism of mussel adhesion to different surfaces has driven the development of advanced dispersing agents with strong adsorption to oxide nanoparticles. The investigation of dopamine, caffeic acid, tiron and other molecules from the catechol family, and various molecules from salicylic acid, gallic acid, and chromotropic acid families revealed their strong adsorption to metal oxide surfaces. The analysis of dispersion and deposition yield data for various materials provided an insight into the influence of molecular structures of the organic dispersants on adsorption mechanisms and EPD efficiency. The adsorbed dispersants imparted new and unique properties to the nanoparticles. Further advancements in the EPD technology were achieved by the use of cationic and anionic dyes such as pyrocatechol violet, celestine blue, alizarin red from the catechol family and alizarin yellow, aurintricarboxylic acid and calconcarboxylic acid from salicylate family and their derivatives. It was discovered that polyaromatic dyes can be used as efficient co-dispersants for oxide materials, carbon nanotubes and graphene for the fabrication of composite films by EPD. Another important breakthrough was the development of film forming dispersants for EPD nanotechnology. New strategies have emerged for the synthesis of non-agglomerated nanoparticles of controlled size, organic fibers and coated particles. The use of new dispersants with strong interfacial adsorption and multifunctional properties has driven the development of advanced composites, containing metal oxide nanoparticles, conductive polymers, carbon nanotubes, graphene, polyelectrolytes and other materials. Colloidal and interface chemistry of new dispersing agents is emerging as a new area of technological and scientific interest.
1. Introduction
Electrophoretic deposition (EPD)1–5 is an attractive technique for the fabrication of thin films and coatings for various applications. EPD involves electrophoretic motion of charged particles in a suspension toward an electrode and deposit formation under the influence of an applied electric field. Cathodic or anodic deposits can be obtained depending on a particle charge. There are excellent reviews, describing deposition mechanisms, kinetics and applications of EPD.6–12Many investigations were focused on the development of charged dispersants for EPD.13–17 Inorganic acids and alkalis were widely used as additives for the charging and electrostatic stabilization of colloidal particles.1,9 The application of this strategy to nanotechnology of inorganic materials presents difficulties related to chemical reactions of acids or alkalis with inorganic nanoparticles. The charging of inorganic particles can be achieved by the adsorption of ions from the solutions of metal salts. However, metal ions, adsorbed on the colloidal particles, usually participate in electrode reactions and incorporate into the deposits as corresponding hydroxides or oxides.1 Another approach involved iodine–acetone reactions, which generated protons for particle charging. Difficulties of this method are attributed to poor suspension stability, and incorporation of iodine into the deposits.1 Another problem is related to chemical incompatibility of acetone with some binder materials. Various polyelectrolytes were successfully used as charging and film forming additives for EPD of inorganic materials.12 This approach has some limitations related to bridging flocculation of small particles.
Other methods of particle dispersion and charging in solvents are based on the use of surfactants, organic acids and alkalis.18 Fundamental studies were focused on the investigation of dispersant adsorption on particle surface, dispersion mechanisms, suspension stability, electrokinetic properties and deposition efficiency.4,18–25
Despite the impressive progress achieved in the development of EPD method, there is a need for simple and versatile techniques for the efficient dispersion and charging of various materials. A critical property of a dispersant is its adsorption on the particle surface. An adsorbed ionic dispersant imparts a charge to the particles and allows for their EPD. However, a non-adsorbed ionic dispersant acts as an electrolyte. It increases the ionic strength of the suspension, decreases the electrostatic repulsion of particles and facilitates agglomeration. The increase in the ionic strength results in increased conductivity of the suspensions, enhanced gas evolution at the electrode and significant pH changes at the electrode surface. The pH increase at the cathode or pH decrease at the anode can lead to decreasing charge or charge reversal of the particles approaching to the electrode surface.1 In this case poor deposit adhesion is usually observed. The increase in electric current in suspensions results in porous films. Moreover, the electromigration of non-adsorbed ionic dispersant and its accumulation at the electrode surface can affect the deposition of particles. Therefore, the development of advanced dispersing agents with strong adsorption on inorganic particles is of paramount importance for the development of EPD and other colloidal techniques.
The adsorption of dispersing agents on inorganic particles is influenced by particle–solvent interactions, solvation effects and dielectric constant (ε) of the solvent. For adsorption governed by electrostatic interactions, the energy of charge interaction in a solvent is decreased by a factor of 1/ε. For the non-covalent interactions, governed by dipole–dipole and dispersion forces,26 the interaction energy decreases by 1/ε2. It is known that water and other liquids lead to the deterioration of performance of many adhesives,26 especially in the presence of ions.
There is a strong need in the development of advanced dispersing agents with strong adsorption to oxide particles. A new avenue for research on the development of efficient dispersing agents with strong adsorption to the particle surface evolved from the investigations of the mechanism of mussel adhesion to various surfaces.26,27 It is known that mussels and other biological species show excellent adhesion to different surfaces in saline water. The very strong and fast adsorption of mussels to different surfaces26 prevents their damage by sea waves. Fundamental studies showed26,28 that strong mussel adhesion involves protein macromolecules, containing catecholic amino acid, L-3,4-dihydroxyphenylalanine (DOPA). The important advantage of the catechol chemistry is its applicability to a large variety of substrates and high binding strength under wet conditions.29 The adhesion mechanism of mussels is attributed to the complexation of metal atoms on material surfaces by OH group of the catechol ligands (Fig. 1). It is known that the catechol group is a powerful complexing agent, despite of its extremely high pKa values.30 It is able to coordinate metal atoms in acidic or basic pH solutions with complete deprotonation of the OH groups.30,31 These studies generated significant interest in the use of DOPA for the fabrication of synthetic polymer adhesives.26,29,32–34 DOPA is now considered as an essential component of many advanced moisture resistant adhesives.26 The fundamental study of mussel adhesion has driven the development of dispersing agents from the catechol family of molecules for the fabrication of stable suspensions of colloidal particles. Investigations showed that other organic molecules with chelating properties, such as molecules from salicylic acid, gallic acid and chromotropic acid family strongly adsorbed on inorganic particles.
 | ||
| Fig. 1 Schematic of mussel adsorption and chemical structure of catechol. | ||
This review describes recent advances in the development of dispersing and charging agents from catechol, gallic acid, salicylic acid and chromotropic acid families for application in EPD. Such organic molecules are of special interest for EPD technology due to their strong adsorption to oxide particles and electric charge. Another advantage of such molecules is their small size, compared to long chain surfactants, and high charge to mass ratio. Special attention is focused on the influence of functional groups, structure and size of the molecules on their adsorption on particle surface, particle dispersion and EPD kinetics. Deposition mechanisms, new colloidal strategies and applications are discussed. Of special interest are new properties of the oxide nanoparticles, which result from dispersant adsorption.
The structures and properties of new dispersants from catechol family and their applications in EPD were described in Section 2 of this review. Other types of dispersants with strong adsorption to inorganic particles were described in Section 3, including organic molecules from gallic acid, salicylic acid and chromotropic acid families. Section 4 is focused on dyes from catechol family, including cationic and anionic dyes. Section 5 describes structures, properties and applications of the dyes from salicylic acid family, which exhibit anionic properties. Special attention is focused on dyes with film forming properties. Recent advances in the development of new dispersing agents open exciting opportunities in nanotechnology, described in Section 6.
2. Adsorption of catechol-based materials and EPD of metal oxide particles
This section describes the chemical structures and adsorption mechanisms of DOPA and other molecules from the catechol family. The applications of cationic and anionic molecules for particle dispersion, charging and EPD of oxide particles are described.2.1. DOPA
DOPA belongs to the catechol family of materials (Fig. 2). The chemical structure of DOPA includes two OH groups bonded to adjacent carbon atoms of the aromatic ring, carboxylic, and amino groups. The total charge of the zwitterionic DOPA molecules is governed by the ionization of different functional groups.35 The functional groups of DOPA are protonated at low pH. Therefore, the positive charge of DOPA at low pH is attributed to the protonated amino group (NH3+).35 The pH increase leads to the deprotonation of carboxylic, phenolic, and amino groups. As a result, DOPA possesses a negative charge at high pH. It was found35 that the amino group is protonated below pH = 10, it reduces the total negative charge of the DOPA molecule at relatively high pH. | ||
| Fig. 2 Structures of molecules from catechol family and other monoaromatic anionic molecules. | ||
Two OH groups of DOPA, bonded to the adjacent carbon atoms of the aromatic ring, allow strong adsorption on different surfaces. It was found that DOPA forms complexes with metal ions, the number of DOPA molecules in the complexes depends on pH.36 As a result, DOPA provides bidentate interfacial interactions on various surfaces, containing metal ions.26,37 In the investigations of DOPA, adsorbed on titanium dioxide in water, the pull-off forces of >500 pN were reported for a single DOPA molecule.26 A single molecular studies of DOPA adsorption showed that oxidation of DOPA reduces the strength of the interaction with metal oxide surface but results in high strength irreversible covalent bond formation with an organic surface.38 It was found that adsorption of DOPA on electrode surface can be used for the fabrication of sensors for chiral electrochemical recognition of biomolecules.39 DOPA was used as a catecholic anchor and initiator for surface-initiated polymerization from metal surfaces to create antifouling polymer coatings.40 Recent advances in the development of adhesives based on DOPA were described in review papers.26,41
The use of DOPA for particle charging for EPD presents difficulties due to zwitterionic properties of this molecule. However, the analysis of DOPA adsorption on various substrates has driven the investigations of other molecules from catechol family. The closest molecular analogues of DOPA are molecules of dopamine and caffeic acid (Fig. 2).
2.2. Dopamine
Dopamine is known as neurotransmitter whose deficiency leads to Parkinson's disease.42 Dopamine, epinephrine (adrenaline) and other molecules from the catecholamine family43,44 have many important functions in the human body. The amino group of dopamine (Fig. 2) can be protonated in solutions of inorganic acids. As a result, dopamine exhibits cationic properties in acidic solutions. In basic solutions, self-polymerization of dopamine was observed, which was investigated for the fabrication of polydopamine coatings. It was found that polydopamine has strong adhesion to different substrates, such as Au, Ag, Pt, Cu, stainless steel, TiO2, SiO2, GaAs, Si3N4, glass and other materials.27,36,38,41,42 Dopamine has generated interest for materials synthesis. The polymerization of dopamine promoted the formation of unstable vaterite phase of CaCO3 from CaCl2 and Na2CO3 solutions, containing dopamine.45 Moreover, dopamine promoted the transformation of vaterite to hydroxyapatite in simulated body fluid solutions.45 Dopamine is of great interest for the development of nanomaterials with advanced properties and their deposition by the EPD method.Several investigations indicated that dopamine adsorption resulted in improved functional properties of nanoparticles. The adsorption of dopamine on magnetic Fe3O4 particles allowed enhanced magnetization and increased superparamagnetic blocking temperature.46,47 It was suggested that dopamine adsorption changed the microstructure of the magnetic dead layer on the particle surface and promoted magnetic ordering in the surface layer.46 Dopamine sensitized TiO2 nanoparticles showed improved photovoltaic properties.48–50 The adsorption of dopamine resulted in changes in structure of particle surface layer and promoted charge transfer48,49,51 at the organic–inorganic interface. The dopamine modified TiO2 was investigated for the application in photoelectrochemical transducers and biosensors.48,50 In another investigation52 dopamine was used as an anchor for the functionalization of nanoparticles using a ‘click’ chemistry method. It has been found53,54 that dopamine strongly adsorbed on ZnO and enhanced luminescence properties. There have been a number of studies which have examined the effect of dopamine adsorption on optical and surface properties of oxides.50,55–57
Dopamine was used for cathodic EPD of MnO2, ZnO and TiO2 (ref. 53, 58 and 59) from suspensions in ethanol or in mixed ethanol–water solvent. Dopamine adsorption allowed improved dispersion of MnO2, ZnO and TiO2. The deposition yield increased with increasing dopamine concentration in the suspensions in the range of 0.01–0.2 g L−1. The increased deposition yield was attributed to increasing DA adsorption on oxide particles and increasing particle charge. The adsorption of dopamine on oxide particles was confirmed by Fourier transform infrared spectroscopy (FTIR) and quartz crystal microbalance (QCM) method.58 The suggested deposition mechanism involved adsorption of dopamine on particle surface, electrophoresis of the charged particles, pH increase at the cathode surface and charge neutralization of the particles at the electrode surface. The charge neutralization promoted deposit formation. It was found that dopamine can be used for the fabrication of composite materials. In the proposed approach dopamine was used as a cationic co-dispersant for individual components. The adsorption of dopamine on oxide particles was based on the chelating mechanism, involving OH groups of the catechol ligand. The adsorption of dopamine on multiwalled carbon nanotubes (MWCNT) involved π–π interactions. The feasibility of the new approach was demonstrated by co-deposition of ZnO and TiO2 (ref. 53) and MnO2 and MWCNT.58 The formation of composite materials was confirmed by electron microscopy,53,58 chemical analysis53 and thermogravimetry methods.58 It was found that Zn/Ti atomic ratio in the deposits increased with increasing Zn/Ti ratio in the suspensions.53 The MnO2–MWCNT composites were used for the fabrication of electrochemical supercapacitors for energy storage.58 The composite electrodes showed significant improvement in capacitance, compared to pure MnO2 due to higher electronic conductivity of the composite. The improved electrochemical performance was achieved by good dispersion of MWCNT in the MnO2 matrix.58 It was found that EPD of ceramic particles, containing adsorbed dopamine as a charging and dispersing agent, can be combined with the EPD of cationic polyelectrolytes for the fabrication of organic–inorganic nanocomposites.59 In this approach dopamine prevented bridging flocculation of TiO2 nanoparticles by the polylelectrolyte.59
2.3. Caffeic acid
Caffeic acid (CA) is important phenolic antioxidant, which is present in many plants and beverages.60 It was found that CA can be used as efficient green corrosion inhibitor for stainless steel.61 Similar to DOPA, the structure of caffeic acid contains two OH groups bonded to adjacent carbon atoms of the aromatic ring (Fig. 2). The anionic properties of CA are related to the dissociation of the carboxylic group (Fig. 2). CA showed strong adsorption on inorganic surfaces, such as stainless steel,61 TiO2,62,63 silica,64,65 zirconia66 and MnO2 (ref. 66) in different solvents. The adsorption of CA on inorganic particles can involve phenolic and carboxylic (Fig. 3) bonding sites.66 Bidentate chelating bonding (Fig. 3a) and bidentate bridging bonding (Fig. 3b) mechanisms33,67 were proposed for CA adsorption.66 The adsorption can involve inner sphere (Fig. 3b) or outer sphere bonding (Fig. 3c), depending on the nature of the adsorbent material.68 The following reaction stoichiometry was proposed66 for bidentate chelating bonding,69,70 involving metal atoms (M), containing surface OH groups, and catechol (L2−H2+):| M–2OH + L2−H2+ → ML + 2H2O | (1) |
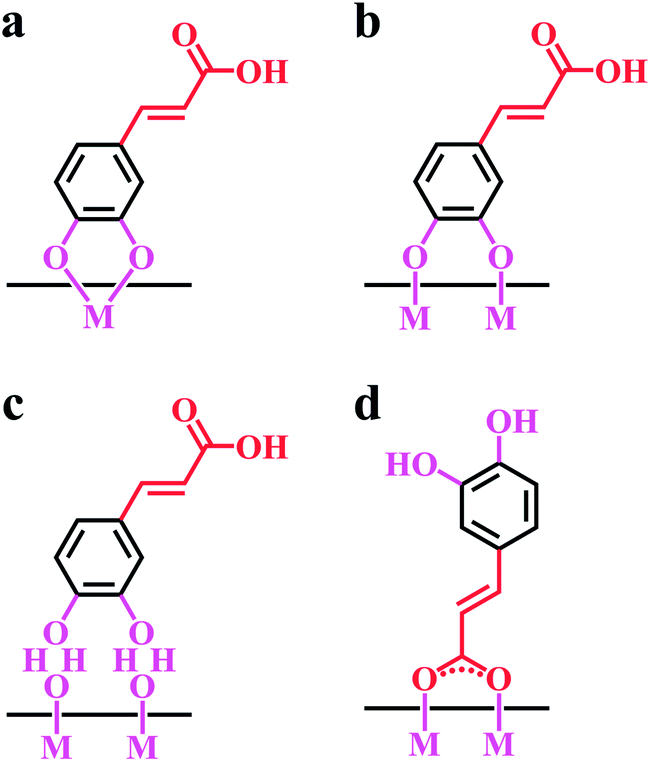 | ||
| Fig. 3 Suggested adsorption mechanisms of caffeic acid: (a) bidentate chelating bonding, (b) bidentate bridging bonding (inner sphere), (c) bidentate bridging bonding (outer sphere) of catechol group, (d) adsorption, involving a carboxylic group. | ||
The bidentate bridging inner-sphere bonding66 is based on the reaction:69,71
| 2M–OH + L2−H2+ → M2L + 2H2O | (2) |
The outer sphere bonding66 is formed as follows:71,72
| 2M–OH + L2−H2+ → (M–OH2+)2–L2− | (3) |
Other mechanisms66 included monodentate bonding, involving one OH group, or mixed monodentate–bidentate bonding.33,73
The adsorption mechanism, involving a carboxylic group74 is shown in Fig. 3d. The investigation of benzoic and phthalic acids, which do not have OH groups bonded to the aromatic ring, provided evidence that carboxylic groups were involved in adsorption of the acids on zirconium, titanium and iron oxides.74,75 Therefore, similar adsorption mechanism can be suggested for the adsorption of CA. Studies of the interactions of CA with different ions in aqueous solutions showed that the bonding mechanism depended on the nature of metal ions. Among the two possible coordination sites, the carboxylate group of CA showed greater complexing power toward Pb(II). However, Al(III) preferentially coordinated the catechol group of CA.76 A spectroscopic study of CA adsorbed on TiO2 indicated that the carboxylic group of CA does not interact with oxide surface.62 It was shown that phenolic groups of CA were involved in adsorption.62
EPD of oxide materials provided important information related to CA adsorption mechanism. TiO2, MnO2 and zirconia suspensions, containing CA, trans-cinnamic acid (TCA), p-coumaric acid (PCA), 2,4-dihydroxycinnamic acid (DCA) were investigated.63,66 The structures of TCA, PCA and DCA are similar to the structure of CA.63 However, TCA does not have OH groups bonded to the aromatic ring (Fig. 2), whereas PCA has only one phenolic OH group. The structure of DCA includes two phenolic OH groups; however, in contrast to CA, they are bonded to non-adjacent carbon atoms of the aromatic ring (Fig. 2). All the organic molecules contain a COOH group.
The suspensions of TiO2 in ethanol were unstable, however the addition of the organic molecules, such as CA, TCA, PCA and DCA resulted in improved suspension stability.63 The best suspension stability was observed for CA containing suspensions, which were stable for more than 1 month. EPD from TiO2 suspension, containing CA resulted in the formation of anodic deposits. In contrast, cathodic deposits were obtained using TCA, PCA and DCA as additives.63 The charge of the particles was governed by the competitive adsorption of dissociated anionic molecules and H+. The preferred adsorption of anionic CA species resulted in the negative charge of the particles, which formed anodic deposits. Preferred adsorption of H+ in the suspensions containing TCA, PCA and DCA additives resulted in the formation of positively charged TiO2 particles, which formed cathodic deposits.63 The FTIR data confirmed the mechanism of CA adsorption, which involves the deprotonation of phenolic OH groups and complexation of Ti atoms. The anodic deposition yield increased with increasing CA concentration in the TiO2 suspensions and deposition time.63
The comparison of the chemical structures of CA, TCA, PCA and DCA and deposition yield data indicated that the interaction of carboxylic or individual OH groups of TCA, PCA and DCA with Ti atoms on the particle surface was weak, compared to catecholate type adsorption of CA. The investigations of zirconia and MnO2 suspensions, containing CA, TCA, PCA and DCA provided further evidence of strong catecholate type adsorption of CA.66 The results indicated that no deposition was achieved from the zirconia suspensions, containing TCA, PCA, and DCA. This was attributed to weak interactions of carboxylic groups or individual OH groups of TCA, PCA, and DCA with particle surface.66 In contrast, the addition of CA to zirconia suspensions allowed the formation of anodic deposits from stable suspensions. The deposition yield was varied by variation of CA concentration in the suspensions and deposition time. The EPD from pure MnO2 suspensions in ethanol showed that nanoparticles of MnO2 were positively charged and allowed the formation of cathodic deposits. The addition of TCA, PCA, and DCA to MnO2 suspensions resulted in decreasing deposition yield; however, no deposition, either cathodic or anodic, was observed at TCA, PCA, and DCA concentrations above 0.05 g L−1. The MnO2 suspensions, containing TCA, PCA, and DCA, were unstable and showed rapid sedimentation immediately after ultrasonic agitation.66 The addition of CA to the MnO2 suspensions resulted in decreasing cathodic deposition yield. No deposition, either cathodic or anodic, was observed at CA concentration of 0.05 g L−1. At higher CA concentrations, anodic deposition was observed. The anodic deposition yield increased with increasing CA concentration. The increase in the CA concentration, deposition voltage and deposition time resulted in increasing deposition yield. The formation of anodic deposits indicated that MnO2 particles were negatively charged at CA concentration above 0.05 g L−1. The charge reversal of the MnO2 particles in suspensions, containing CA, resulted from strong CA adsorption on the particles. The adsorption of CA on zirconia and MnO2 was confirmed by FTIR studies of the films, deposited by EPD.66
The analysis of EPD yield data for zirconia and MnO2 powders annealed at different temperatures provided additional information related to the adsorption mechanism. The deposition yield data for zirconia and MnO2 annealed at different temperatures, indicated that changes in crystallinity had no appreciable effect on CA adsorption and EPD of both materials.66 It is in this regard that the chemical composition of the metal hydroxide/oxide, rather than the crystal structure, was found to determine the structure of the ligand–surface complex.77 The deposition yield decreased with increasing annealing temperature. It was found that annealing resulted in the condensation of surface OH groups and water release. The removal of the surface OH group resulted in reduced adsorption of CA, which, in turn, led to reduced particle charge and decreased deposition rate. The result indicated that CA formed outer-sphere complexes (Fig. 3) with MnO2 and zirconia.66 This finding correlated with the results of another investigation68 which showed that catechol forms primarily outer-sphere complexes with MnO2.
The strong adsorption of CA on particle surface paves the way for the CA application for synthesis of nanoparticles by chemical precipitation methods. It was found that adsorbed CA prevented agglomeration of zirconia nanoparticles during synthesis. The use of CA for synthesis, dispersion and charging of TiO2, MnO2 and zirconia nanoparticles allowed the EPD of uniform and dense deposits on various conductive substrates.63,66 It was found that CA can be used as a co-dispersant for anodic deposition of composite films. Proof-of-concept was demonstrated by the deposition of composite MnO2–zirconia films. Electron microscopy and energy dispersive spectroscopy investigations showed that film composition can be varied by the variation of concentration of MnO2 and zirconia in the suspensions.66
2.4. 3,4-Dihydroxybenzoic acid, 3,4-dihydroxyphenylacetic acid and 3,4-dihydroxyhydrocinnamic acid
Fig. 2 shows chemical structures of 3,4-dihydroxybenzoic acid (DHB), 3,4-dihydroxyphenylacetic acid (DHP) and 3,4-dihydroxyhydrocinnamic acid (DHC) molecules, which belong to the catechol family. The length of hydrocarbon chains increases in the order DHB < DHP < DHC. The anionic properties of the molecules are related to the carboxylic groups. Molecules of DHB, DHP and DHC were used as charging and dispersing agents for EPD of different oxide materials from suspensions in ethanol. The colloidal stability and deposition yield data for DHB and DHC were compared with the data for other molecules, which have a similar structure (Fig. 2). Benzoic acid and 3-phenylpropionic acid (Fig. 2) do not have phenolic OH groups, whereas 4-hydroxybenzoic acid and 3-(4-hydroxyphenyl) propionic acid have only one phenolic OH group. The structure of 3,5-dihydroxybenzoic acid includes two phenolic OH groups; however, in contrast to DHB, they are bonded to non-adjacent carbon atoms of the aromatic ring (Fig. 2). All the organic molecules contain a COOH group. The investigations of MnO2 suspensions showed that only DHB, DHP and DHC molecules allowed good suspension stability and film formation by anodic EPD. The comparison of the molecular structures, deposition yield and FTIR data indicated that strong adsorption of DHB, DHP and DHC on MnO2 is related to the complexation of Mn atoms by the catechol ligands.78 The deposition yield increased in the order of DHB < DHP < DHC. The increase in the deposition yield correlates with increase in the length of the hydrocarbon chains of the corresponding molecules. Such increase was attributed to the improved dispersion, provided by adsorbed molecules with longer hydrocarbon chain.78 The EPD method allowed the formation of uniform deposits, containing non-agglomerated nanoparticles. The deposition yield measurements showed that the amount of deposited material can be varied by the variation of dispersant concentration in the solutions and deposition time. The use of DHB as a dispersant for synthesis of MnO2 particles and their EPD allowed the formation of uniform films, containing non-agglomerated MnO2 nanoparticles. The MnO2 films, prepared by EPD can be used for applications in energy storage devices, such as supercapacitors and batteries.78Recent studies showed that DHB, DHP and DHC are promising dispersants for EPD of other oxide materials. Of particular interest are investigations, which revealed strong DHB and DHP adsorption on TiO2 nanoparticles.31,79 The results of FTIR studies showed that adsorption involved the formation of inner-sphere charge-transfer complex.31 The adsorption of DHB resulted in significant change in optical properties and effective band gap of TiO2. The formation of bidentate binuclear complexes resulted in restoration of six-coordinated octahedral geometry of surface Ti atoms.31 It was found that TiO2 nanoparticles modified with DHB are promising materials for photovoltaic applications. In another investigation80 DHC was used for the synthesis and dispersion of Fe3O4. The adsorption of DHC on Fe3O4 during synthesis allowed the fabrication of nanoparticles of controlled size, which showed a saturation magnetization similar to that of the bulk material. The adsorption mechanism involved complexation of Fe atoms with catechol. It was found that COOH group of DHC was exposed to the surrounding water and promoted good nanoparticle dispersion in water.80 Zeta-potential measurements showed that dissociated COOH group of adsorbed DHC imparted a negative charge to the particles. The DHC modified Fe3O4 particles were investigated for biomedical applications.
2.5. Tiron
Tiron (4,5-dihydroxy-1,3-benzenedisulfonic acid disodium salt) belongs to the catechol family of materials (Fig. 2). The anionic properties of tiron are attributed to two SO3− groups. This material is of special interest for EPD technology and other electrochemical film deposition techniques. Tiron showed strong adsorption on alumina,81–84 titania,85–87 CuO,88 SnO,89,90 Fe3O4,47 α-Fe2O3,91 hydroxyapatite,92 yttria stabilized zirconia93,94 and cordierite-based glass–ceramics.95 It was shown that tiron forms inner sphere complexes with metal atoms on material surface.85,88,96 The adsorption of tiron was studied by electrophoretic experiments and FTIR studies.88,95 The use of tiron as a dispersant allowed the formation of stable suspensions in aqueous and mixed water–ethanol solvents.47,86,92,93 Electrophoretic mobility measurements showed that adsorbed tiron imparted a negative charge to oxide particles.81,92,94 The adsorption of tiron on inorganic particles resulted in shift of their isoelectric points to lower pH and increase in absolute value of zeta-potential.81,92,93 The comparison of tiron with other advanced dispersants for alumina suspensions showed that good suspension stability can be achieved at lower tiron concentration, compared to other dispersants.84 This can be attributed to good adsorption of tiron on particle surface and high charge to mass ratio of this dispersant. Well dispersed slurries with a solid content as high as 50% were obtained.92 The use of tiron for synthesis of inorganic oxides allowed controlled particle growth.91 Tiron was used as a reducing and dispersing agent for the fabrication of gold nanoparticles by a chemical precipitation method.97 The investigations of tiron adsorption on different materials provided a scientific basis for application of tiron for EPD.Several investigations were focused on EPD of TiO2 films from suspensions in water or mixed water–ethanol solvent.85–87 The analysis of TiO2 suspension in a mixed water–ethanol solvent showed that tiron adsorption on TiO2 particles increased in the presence of ethanol. This was attributed to poor solubility of tiron in ethanol.86 The use of a mixed solvent allowed reduced gas evolution during EPD and improved deposit adhesion. It was found that tiron concentration of 0.05 wt% in the suspensions was sufficient for film formation by EPD.85 Best results were obtained for the tiron concentration in the suspensions in the range of 0.1–0.3 wt% and TiO2 content of 15 wt%. The deposit mass increased linearly with increasing deposition time during the galvanostatic deposition85 from TiO2 suspensions in a mixed water–ethanol solvent. The deposition method allowed controlled EPD of thick coatings with thickness in the range of 0.5–5 mm. It was shown that layers with green density of 60% and narrow pore size distribution can be deposited. The pore size distribution was centered at a pore size of 0.15 μm. As a result, the deposits showed good sintering behavior.85 The EPD method allowed the fabrication of uniform deposits on high surface area stainless steel substrates.87 The coatings prepared by EPD are promising for catalytic and photovoltaic applications.
3. Applications of molecules from gallic acid, salicylic acid and chromotropic acid families for EPD
This section describes the chemical structures of molecules from gallic acid, salicylic acid and chromotropic acid families, their basic adsorption mechanisms and applications for EPD of metal oxide particles.3.1. Gallic acid
Fig. 4 shows a chemical structure of gallic acid (GA), which includes three OH groups bonded to adjacent carbon atoms of the aromatic ring and COOH group. Similar to materials from catechol family, the OH groups provided strong adsorption of GA on various oxides. The anionic properties of GA are related to COOH group. The investigation of surface complexation of GA on TiO2 showed that GA chemisorbs by complexation through two OH groups.98 The third OH and COOH did not influence much the stability of the surface complex. In another investigation99 the adsorption of GA on MnO2 was compared with that of benzoic acid using QCM method. It was found that GA acid strongly adsorbed on MnO2, whereas weak adsorption of benzoic acid was observed.99 The comparison of chemical structures of benzoic acid (Fig. 2) and GA (Fig. 4) indicated that phenolic OH groups of GA were involved in adsorption. GA showed strong adsorption on alumina100 and allowed the fabrication of stable suspensions with volume concentration of the alumina nanoparticles of 40%. It was shown that high concentration of suspension, achieved using GA, facilitates the fabrication of a wide range of products.100 Gallic acid showed strong adsorption on other inorganic materials, such as Fe3O4,101 MnO2,78,99,102 kaolin,103 ZrB2,104 Au105 and provided their efficient dispersion. The analysis of adsorption mechanism provided further evidence of complexation of different atoms on material surface with OH groups of GA. There results paved the way for EPD of various oxides and inorganic composites using GA as a charging and dispersing agent. | ||
| Fig. 4 Structures of materials from gallic acid, salicylic acid and chromotropic acid families. | ||
The use of GA as a dispersing and charging agent allowed improved stability of MnO2 suspensions and anodic films were obtained on various conductive substrates.78,99,102 The comparison of the electrophoretic deposition yield data for MnO2 and TiO2 suspensions containing GA and benzoic acid and FTIR data confirmed the importance of phenolic OH groups for GA adsorption. The dissociation of benzoic acid and GA resulted in the competitive adsorption of H+ and anionic benzoic acid or GA species on particle surface. It was found that MnO2 nanoparticles in ethanol were positively charged and formed cathodic deposits.99 The addition of benzoic acid (Fig. 2) resulted in reduced cathodic EPD yield. In contrast, the addition of GA (Fig. 4) resulted in charge reversal and formation of anodic deposits.99 The addition of benzoic acid to TiO2 suspensions allowed the formation of cathodic deposits. In contrast, anodic deposition was achieved using GA. The addition of GA to TiO2 and MnO2 suspensions led to increased deposition yield and improved suspension stability. It was found78 that efficient EPD of MnO2 can be achieved at GA concentration as low as 0.05–0.1 g L−1. The comparison of the EPD yield data for GA and DHB at low dispersant concentrations78 indicated that GA, containing three OH groups (Fig. 4), is a more efficient dispersing and charging additive than DHB, containing two OH groups (Fig. 2). The EPD yield from MnO2 and TiO2 suspension was varied by the variation of deposition time, voltage and GA concentration.102 The thickness of the deposits was varied in the range of 0–10 μm. The method allowed the fabrication of uniform films with packing density in the range of 42–51%.
It was found102 that GA can be used as a co-dispersant for MnO2 and TiO2 for EPD of MnO2–TiO2 composites. The Mn/Ti ratio was varied by the variation of concentration of MnO2 and TiO2 in the suspensions. The use of GA as a co-dispersant for MnO2 and MWCNT allowed the fabrication of MnO2–MWCNT composites.99 It was suggested that the adsorption of GA on MWCNT is attributed to π–π interactions. The adsorbed GA provided a negative charge for EPD of MWCNT. The formation of MnO2–MWCNT composites was confirmed by electron microscopy and thermogravimetric analysis.99 Electron microscopy showed that the use of GA as a co-dispersant allowed uniform distribution of MWCNT in the MnO2 matrix. The composite materials, prepared by EPD were investigated for application in electrodes of electrochemical supercapacitors.99
3.2. Salicylic acid and related materials
Fig. 4 shows a chemical structure of salicylic acid (SA), which includes OH and COOH groups, bonded to adjacent carbon atoms of the aromatic ring. The anionic properties of SA are related to COOH group. SA showed adsorption on TiO2,31,74,75,102,106,107 hematite,108 ZrO2,75 Al2O3,75,81,109 MnO2 (ref. 110) and Ta2O5.75 The suggested adsorption mechanisms31,74,75,110 involved bidentate chelating bonding and bidentate bridging bonding, including inner sphere and outer sphere bonding (Fig. 5A). The adsorption of SA on TiO2 resulted in improved optical and charge transfer properties.31 Investigations of other molecules from salicylate family (Fig. 4) showed their adsorption on various oxides, including adsorption of 2,5-dihydroxybenzoic acid on TiO2,31 5-sulfosalicylic acid (SSA) on Fe3O4,111 α-Fe2O3,112 Al2O3 (ref. 81) and TiO2 (ref. 113 and 114) and 2,6-dihydroxybenzoic acid on TiO2.114 | ||
| Fig. 5 Suggested adsorption mechanisms of (A(a–c)) salicylic acid and (B(d–f)) chromotropic acid: (a and d) bidentate chelating bonding, (b and e) bidentate bridging bonding (inner sphere), (c and f) bidentate bridging bonding (outer sphere). | ||
EPD of MnO2 and TiO2 was performed from suspensions in ethanol using SA as a charging and dispersing agent.102,110 The addition of SA to the MnO2 suspensions resulted in improved suspension stability and allowed anodic EPD of MnO2 films.110 This investigation confirmed the mechanism of SA adsorption, involving COOH and OH groups of SA, bonded to adjacent carbon atoms of the aromatic ring. The TiO2 suspensions containing SA were unstable and no EPD was observed from such suspensions.102 In contrast, anodic deposition was observed from TiO2 suspensions containing sodium salicylate. The deposition rate increased with increasing concentration of sodium salicylate, showed a maximum at concentration of 0.2 g L−1 and then decreased. It was suggested102 that the electrokinetic behavior of the TiO2 particles in the suspensions containing SA or sodium salicylate was governed by the competitive adsorption of anionic salicylate and cationic H+ or Na+. The formation of anodic deposits from the suspensions, containing sodium salicylate, indicated that TiO2 particles were negatively charged owing to the preferred adsorption of salicylate anions.
Anodic EPD of TiO2 was also performed using SSA and 2,6-dihydroxybenzoic acid.114 The use of 2,6-dihydroxybenzoic acid allowed higher deposition yield and improved suspension stability. The difference was related to SO3− group of SSA, which promoted mutual electrostatic repulsion of TiO2 particles, containing adsorbed SSA, and non-adsorbed anionic SSA accumulated at the electrode surface.114 Such electrostatic repulsion was detrimental for deposit formation at the electrode.
The chemical structures of 2,3-dihydroxybenzoic acid and 2,3,4-trihydroxybenzoic acid (Fig. 4) allow two different bonding mechanisms: salicylate bonding, involving adjacent COOH and OH groups, or catecholate bonding, involving two adjacent OH groups. EPD of MnO2 was performed from the suspensions, containing 2,3,4-trihydroxybenzoic acid.110 The suspensions showed good stability and anodic deposition was achieved at 2,3,4-trihydroxybenzoic acid concentrations above 0.05 g L−1. The deposition yield from MnO2 suspensions, containing 2,3,4-trihydroxybenzoic acid was significantly higher compared to that achieved form the suspension, containing SA additive.110 The amount of the deposited materials was controlled by the variation of the additive concentration, deposition time and voltage. The use of 2,3,4-trihydroxybenzoic acid as a dispersant for the synthesis of MnO2 nanoparticles and their EPD allowed the formation of uniform films, containing non-agglomerated MnO2 nanoparticles. It was found that 2,3,4-trihydroxybenzoic acid can be used as a co-dispersant for MnO2 and MWCNT for the fabrication of composite MnO2–MWCNT films.110 The EPD method was further developed for the impregnation of Ni-plaque current collectors with MnO2–MWCNT active material for application in electrochemical supercapacitors. The composite electrodes prepared by EPD showed high specific capacitance at high material loadings, good capacitance retention at high charge–discharge rates and low impedance.110 In another investigation114 2,3-dihydroxybenzoic acid was used for EPD of TiO2. The deposition yield measurements showed that the deposit mass increased rapidly with increasing 2,3-dihydroxybenzoic acid concentration in the range of 0–0.1 g L−1 and only relatively small variations in the deposit mass were observed at higher concentrations in the range of 0.1–1.0 g L−1. The use of 2,3-dihydroxybenzoic acid allowed the fabrication of adherent and uniform deposits from stable suspensions, contacting well dispersed TiO2 particles.114
Similar to SA, the chemical structure of 2-hydroxy-1-naphthoic acid includes COOH and OH groups, which were beneficial for adsorption on particle surface. Fig. 6 shows deposition yield, achieved from MnO2 suspension as a function of 2-hydroxy-1-naphthoic acid concentration in the suspension. The cathodic deposition yield of MnO2 decreased with increasing additive concentration in the range of 0–0.07 g L−1 and no cathodic deposition was observed at higher concentrations. However, anodic deposition was observed at higher additive concentrations. The deposition rate increased with increasing concentration of 2-hydroxy-1-naphthoic acid additive. The charge reversal indicated strong adsorption of the anionic additive on the particle surface. The adsorption mechanism involved the salicylate type of bonding.
 | ||
| Fig. 6 Deposit mass versus 2-hydroxy-1-naphthoic acid concentration in 4 g L−1 MnO2 suspension at a deposition voltage of 40 V (distance between electrodes 15 mm) and deposition time of 3 min: (a) cathodic deposition, (b) anodic deposition. | ||
3.3. Chromotropic acid
The interest in chromotropic acid (CHR) for EPD is related to complexing properties of this molecule (Fig. 4) and electric charge. It is known that CHR forms complexes with different metal ions and shows good adsorption on different surfaces.99,115–118 The adsorption mechanisms can be attributed to bidentate chelating bonding or bidentate bridging bonding, including inner sphere and outer sphere bonding (Fig. 5B), which involves two OH groups of CHR. The negative charge of CHR is related to two SO3− groups (Fig. 4). CHR was investigated for EPD of MnO2 from suspensions in ethanol.99 The addition of CHR to MnO2 suspensions resulted in charge reversal of MnO2 particles from positive charge to negative charge due to adsorption of anionic CHR. The anodic deposition rate increased with increasing CHR concentration in the range of 0.05–0.3 g L−1. The decrease in the deposition rate at higher concentrations was related to increasing ionic strength of the suspensions. High deposition rate was obtained from 4 g L−1 MnO2 suspensions at CHR concentrations in the range of 0.2–0.3 g L−1. The use of CHR allowed the fabrication of uniform and adherent deposits from stable suspensions. The deposition yield was well controlled by variation of deposition voltage, time and CHR concentration.4. Applications of dispersing agents from catechol family of dyes for EPD
The section is focused on dyes from catechol family. Adsorption mechanisms, particle charging and EPD of oxide particles using cationic and anionic dyes are described.4.1. Cathodic EPD using celestine blue dye
Organic dyes are of special interest for EPD due to their important properties and possibility of functionalization of inorganic nanoparticles. The larger size of the dyes, compared to that of monoaromatic molecules (Fig. 2), is beneficial for electrosteric stabilization of nanoparticles in suspensions. Moreover, organic dyes have generated significant interest for the dispersion of carbon nanotubes and graphene. Therefore, organic dyes are promising co-dispersants for co-deposition of various materials.Fig. 7 shows a chemical structure of cationic celestine blue (CB) dye, which is under investigations for application in solar cells and sensors.119,120 CB belongs to the catechol family of materials. The positive charge of CB makes this molecule important for application as a charging and dispersing agent for cathodic EPD of metal oxides. The larger size of CB, compared to that of dopamine, is beneficial for the electrosteric dispersion. It is important to note that dopamine has no charge and must be protonated in acidic solutions for application in EPD. The dopamine is unstable in basic solutions due to self-polymerization. In contrast, the dissociation of CB allows the formation of stable solutions of cationic CB species.
 | ||
| Fig. 7 Chemical structure of cationic celestine blue dye. | ||
Similar to other materials from the catechol family, CB showed strong adsorption on various oxide materials. It was found that adsorbed CB provided improved suspension stability and imparted a positive charge to the particles. Various oxide materials were deposited by cathodic EPD, such as TiO2, BaTiO3, MnO2, Mn3O4, Y2O3. Fig. 8A shows deposition yield from TiO2 suspensions in ethanol as a function of CB concentration in the suspensions. The addition of CB to the suspensions resulted in rapid increase in the deposition rate in the range of 0–0.1 g L−1. At higher concentrations, the deposition yield increased gradually with increasing CB concentration. The addition of CB resulted in the formation of stable suspensions. The deposition yield increased with increasing deposition time, indicating the formation of deposits of different mass (Fig. 8B). Fig. 9 shows a microstructure of a BaTiO3 deposit, prepared by EPD. The deposit was relatively dense and exhibited a small porosity, which can be attributed to packing of BaTiO3 particles. Fig. 10 shows the microstructure of a TiO2 film, prepared using CB as a dispersing and charging agent. The film contained nanoparticles of TiO2, the pore size was below 100 nm. CB can be used for EPD of other materials, such as halloysite nanotubes (Fig. 11). The EPD coatings, containing hollow halloysite nanotubes are currently under investigation for controlled drug delivery applications. Moreover halloysite can improve mechanical and flame retardant properties of composite materials.121,122
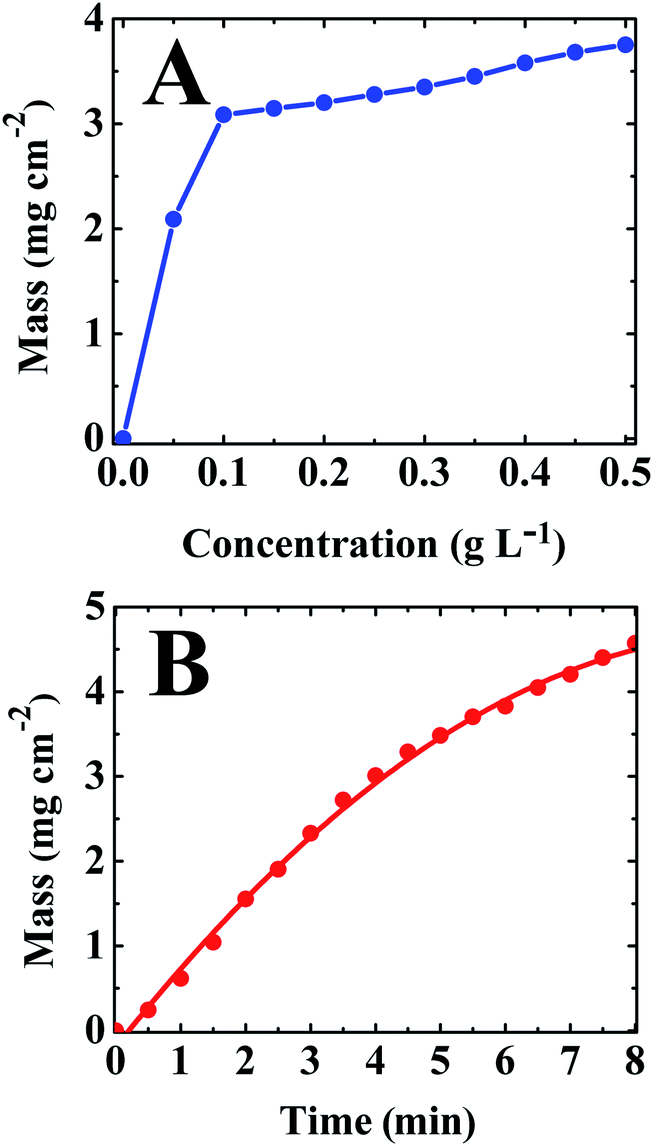 | ||
| Fig. 8 Deposit mass for films prepared from 10 g L−1 TiO2 suspension in ethanol at a deposition voltage of 40 V (distance between electrodes of 15 mm): (A) versus celestine blue concentration at a deposition time of 5 min and (B) versus deposition time at celestine blue concentration of 0.5 g L−1. | ||
 | ||
| Fig. 9 SEM image of BaTiO3 film prepared using celestine blue dispersant. | ||
 | ||
| Fig. 10 SEM image of TiO2 film prepared using celestine blue dispersant. | ||
 | ||
| Fig. 11 SEM image of halloysite nanotube film prepared using celestine blue dispersant. | ||
4.2. Anodic EPD using alizarin red, pyrogallol red and pyrocatechol violet dyes
Fig. 12 shows chemical structures of anionic alizarin red (AR), pyrogallol red (PR) and pyrocatechol violet (PV) dyes, which were investigated for application in EPD. The anionic properties of the dyes are related to their SO3− groups. The structures of the AR, PR and PV dyes include a catechol ligand. AR showed strong adsorption on various materials such as Mn3O4,123 TiO2,124,125 clays,126,127 hydroxyapatite.128 FTIR studies showed that adsorption of AR involved complexation of metal atoms on particle surface by two adjacent OH groups or adjacent OH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups of AR (Fig. 12).125,128 The investigation of PR adsorption129 on TiO2 is important for the development of solar cells. The catechol complexing ligand of PR was involved in the formation of charge transfer complex of TiO2 and PR. The excitation of PR dye adsorbed on TiO2 particles allowed efficient electron injection into the conduction band of TiO2. The discovery of efficient electron injection dynamics in the PR–TiO2 system has generated significant interest in the fabrication of PR–TiO2 hybrid materials. PV adsorbed on various metal oxides and formed charge transfer complexes through the catechol ligand with a bidentate bonding.130,131 The strong interaction of PV with metal oxides allowed the fabrication of hybrid films130 with improved charge transfer between PV and inorganic components.
O groups of AR (Fig. 12).125,128 The investigation of PR adsorption129 on TiO2 is important for the development of solar cells. The catechol complexing ligand of PR was involved in the formation of charge transfer complex of TiO2 and PR. The excitation of PR dye adsorbed on TiO2 particles allowed efficient electron injection into the conduction band of TiO2. The discovery of efficient electron injection dynamics in the PR–TiO2 system has generated significant interest in the fabrication of PR–TiO2 hybrid materials. PV adsorbed on various metal oxides and formed charge transfer complexes through the catechol ligand with a bidentate bonding.130,131 The strong interaction of PV with metal oxides allowed the fabrication of hybrid films130 with improved charge transfer between PV and inorganic components.
 | ||
| Fig. 12 Chemical structures of anionic dyes from catechol and salicylate family. | ||
AR, PR and PV were used for EPD of TiO2 and MnO2 (ref. 114, 125, 131 and 132) from suspensions in ethanol. The particles of TiO2 and MnO2 were deposited anodically from stable suspensions on different substrates. PR was also used as a dispersant for the synthesis of MnO2. The adsorption of PR on the particle surface limited particle growth related to Ostwald ripening, resulting in reduced particle size and reduced agglomeration.132 The deposition yield was varied by variation of dye concentration in suspension, deposition time and voltage.114,125,131,132 EPD resulted in uniform films with typical thickness in the range of 0–10 μm. The analysis of FTIR data confirmed that the catechol complexing ligands of the dyes governed the adsorption mechanism. The nanostructured films of MnO2 were used for the fabrication of supercapacitor electrodes,132 which showed a specific capacitance of 409 F g−1.
The use of AR, PR and PV114,125 as charging and dispersing agents for EPD of TiO2 offers advantages for photovoltaic applications. Previous work on EPD of TiO2 involved different charging agents and the deposited films were impregnated with dye solutions.133,134 This approach has some inherent problems related to film impregnation with dyes, competitive adsorption of the dispersing agents and dyes. Another problem is related to maintaining the film integrity during impregnation. Such problems were avoided using AR, PR and PV114,125 dyes as charging and dispersing agents, which adsorbed on TiO2 in the bulk of suspensions and incorporated into the deposits.
Recently, a new strategy has emerged for the fabrication of composite metal oxide–carbon nanotube films. In this strategy, PV was used as a dispersant for the dispersion of MWCNT. The dispersion mechanism in aqueous and non-aqueous suspensions114,131,135 involved adsorption of PV on MWCNT, governed by π–π interactions. It was found that pristine MWCNT can be efficiently dispersed and deposited by EPD. The dispersion was achieved at relatively low PV concentrations. It was found that PV can be used as a co-dispersant for oxide materials and MWCNT. This approach was used for the fabrication of composites, such as TiO2–MWCNT114 and MnO2–MWCNT.131 The method allowed good control of deposit composition and microstructure, which resulted in enhanced electrochemical performance.131
The possibility of independent deposition and co-deposition of oxide particles and MWCNT offers processing advantages, compared to other EPD methods which are based on the fabrication of building blocks, containing oxide particles and carbon nanotubes and deposition of the building blocks.136 Such building blocks were formed due to the opposite surface charges of oxide particles and carbon nanotubes.136 The method required several steps and introduced inherent problems, such as heterocoagulation of the components in the bulk of the suspensions, the dispersion and deposition of relatively large blocks and control of total charge of the blocks.136
5. Anodic EPD using dyes from salicylic acid family
This section is focused on dyes from salicylic acid family. Chemical structures, adsorption mechanisms and applications of anionic dyes for EPD are described. New approach is based on the use of dyes with film forming properties. EPD methods are described for deposition of dyes and metal oxide films.5.1. Alizarin yellow, aurintricarboxylic acid and calconcarboxylic acid
Fig. 12 shows chemical structures of alizarin yellow (AY), aurintricarboxylic acid (ATH) and calconcarboxylic acid (CCH), which belong to the salicylic acid family of molecules, containing adjacent COOH and OH groups. The negative charge of AY and ATH is related to their COO− groups. The structure of CCA includes anionic COO− and SO3− groups.AY was used for EPD of ZnO and TiO2 from suspensions in ethanol.53,125 The nanoparticles of ZnO were positively charged in pure ethanol and formed cathodic deposits.53 The addition of anionic AY resulted in charge reversal, attributed to adsorption of AY. Anodic films were obtained at AY concentration above 0.09 g L−1. The deposition yield increased significantly in the range of 0.09–0.21 g L−1, showed a maximum and decreased at higher concentrations due to increased ionic strength of the suspensions. A similar maximum was observed in the deposition yield versus AY concentration dependence for TiO2 suspensions.125 The FTIR studies of ZnO53 and TiO2 (ref. 125) deposits confirmed AY adsorption.
Detailed studies of ATH137,138 showed that this dye forms complexes with various ions and can be adsorbed on various surfaces. ATH was used for anodic EPD of MnO2 (ref. 78) in ethanol. The addition of ATH allowed the fabrication of stable suspensions. High deposition rate was achieved at ATH concentrations of 0.05–0.1 g L−1 and no significant variation in the deposition yield was observed at higher concentrations. The use of ATH allowed the formation of uniform deposits, containing non-agglomerated nanoparticles at controlled deposition rate.
CCH is of special interest for EPD of oxide materials and composites.139 The suspensions of MnO2 and MWCNT in ethanol, containing CCH showed excellent stability. CCH was used as a co-dispersant for anodic co-deposition of MnO2 and MWCNT and fabrication of MnO2–MWCNT composites.139 The results of deposition yield measurements for CCH, analysis of FTIR data, comparison of the experimental data for CCH and other dyes with different chemical structures provided insight into the mechanism of CCH adsorption on MnO2. The results indicated that adsorption of CCH on MnO2 particles involved salicylate type bonding.139 It was found that CCH and other polyaromatic materials from salicylate family are more efficient dispersing agents for EPD, compared to monoaromatic salicylates. Further research resulted in the development of advanced salicylate-type dispersants with film forming properties.
5.2. Film forming dyes: aluminon and pamoic acid sodium salt
Aluminon (AT(NH4)) and pamoic acid sodium salt (PANa) are water soluble dyes (Fig. 12) from the salicylate family, which exhibit strong chelating properties.137,138,140,141 The chelating properties of the dyes promoted their adsorption on various metal oxides and allowed good dispersion of oxide particles in water.84,94,142–145Aluminon and PANa exhibit interesting properties, which are of special interest for EPD. It is important to note that in contrast to AT(NH4) and PANa, the aurintricarboxylic acid (ATH) and pamoic acid (PAH) are insoluble in water. Recent studies showed that thin films of ATH can be obtained from aluminon solutions by EPD.125,146 The deposition mechanism involved the surface pH decrease at the anode due to the reaction:
| 2H2O → O2 + 4H+ + 4e− | (4) |
Electric field provided electrophoretic motion of anionic AT− toward the anode, where the charge neutralization of COO− groups of AT− resulted in the formation of water insoluble ATH.
| AT− + H+ → ATH | (5) |
A similar mechanism allowed EPD of water insoluble PAH films from PANa solutions:
| PA− + H+ → PAH | (6) |
Fig. 13 shows film mass versus deposition time for ATH deposition from aqueous aluminon solution. The increase in film mass indicates continuous film growth. The decrease in deposition rate with time can be attributed to voltage drop in the growing film. Fig. 14 shows a typical SEM image of a film cross section (fracture). The SEM image indicates that the method allows the formation of relatively uniform films. ATH films were obtained at a constant voltage or potentiodynamic conditions. Film thickness was typically in the range of 0–2 μm.146 The atomic force microscopy studies showed that the root mean square surface roughness of the film was 3.5 nm. The surface roughness of the films was attributed to the gas evolution at the electrode surface during electrodeposition.146 The deposition mechanism was confirmed by FTIR studies.146 In contrast to relatively dense ATH films, the PAH films showed a porous fibrous morphology (Fig. 15A). The SEM image at high magnification (Fig. 15B) indicated that the nanofiber diameter was in the range of 20–50 nm. The fibrous microstructure of such films can result from π–π interactions of the dyes.147
 | ||
| Fig. 13 Film mass measured using QCM for ATH film on a gold coated quartz electrode with area of 0.2 cm2 at a deposition voltage of 4 V (distance between electrodes 10 mm), deposited from 0.1 g L−1 AT(NH4) solution in water. | ||
 | ||
| Fig. 14 Cross section of ATH film (F) on a platinized silicon wafer substrate (S), deposited from 1 g L−1 AT(NH4) aqueous solution at a deposition voltage of 20 V (distance between electrodes 15 mm) during 4 min, arrows show film and Pt layer. | ||
 | ||
| Fig. 15 (A and B) SEM images of PAH film at different magnifications. | ||
The electrochemical reactions of aluminon and PANa and the formation of corresponding water insoluble acids with film forming properties are beneficial for EPD of oxide particles. In the EPD method, oxide particles must be well dispersed and charged in the bulk of the suspensions. However, the particles must coagulate at the electrode surface to form a film. It was found1 that electrophoresis and particle accumulation at the electrode surface do not necessarily result in deposition. The mutual electrostatic repulsion of the particles accumulated at the electrode surface can prevent their deposition.1 It was found that the electrostatic repulsion of particles, containing adsorbed aluminon or PANa, is avoided due to their charge neutralization in anodic reactions.125,146
In many investigations, the bath formulations for EPD included binders, which allowed the formation of adherent and crack free deposits.1,9,10 Charged ceramic particles provided electrophoretic transport of the adsorbed polymer binder material to the electrode surface. In this strategy, some problems are related to competitive adsorption of the dispersant and binder on particle surface and bridging flocculation of the nanoparticles by the polymer binder macromolecules. Such problems can be avoided using aluminon or PANa dispersants, which exhibit binding and film forming properties.125,146 The formation of water insoluble ATH and PAH binders is important for film integrity and adhesion. Fig. 16 shows typical SEM image of a TiO2 film, prepared using aluminon. The EPD method allowed the fabrication of adherent and crack free films of various materials such as TiO2, MnO2 and Al2O3 using aluminon and PANa as charging and dispersing agents. The investigations of electrophoretic mobility of Al2O3 particles showed that the addition of aluminon resulted in a shift of the isoelectric point by two pH units in the acidic direction, compared with a suspension without dispersant.142 Moreover, the addition of aluminon resulted in significant increase in the magnitude of the negative zeta potential at basic pH values. It was found142 that the use of aluminon as a dispersant allowed homogeneous particle packing in green bodies with green density >67%.
 | ||
| Fig. 16 SEM image of a cross section of a film prepared from 1 g L−1 AT(NH4) aqueous solution, containing 10 g L−1 TiO2, at a deposition voltage of 10 V (distance between electrodes 15 mm), F – film, S – platinised silicon wafer substrate, arrows show film thickness and Pt layer thickness. | ||
The investigation of the EPD yield versus aluminon concentration in the 10 g L−1 TiO2 suspension125 indicated that the deposition yield increased with increasing aluminon concentration and showed a maximum at a concentration of 0.25 g L−1. The deposition yield was nearly constant in the concentration range of 0.5–1 g−1. The increase of the deposition yield was attributed to increasing adsorption of aluminon on TiO2 particles and increasing particle charge. In another investigation143 the increase in electrophoretic mobility of Al2O3 particles with increasing concentration of aluminon was reported. It was found that the increase in aluminon concentration above the optimum concentration, corresponding to maximum adsorption, resulted in reduced suspension stability and increased viscosity94,143 of Al2O3 and ZrO2 suspensions. The increase in concentration of non-adsorbed dispersant resulted in screening of the electrostatic repulsion of the colloidal particles. The screening effect of non-adsorbed aluminon can explain the maximum in the dependence of TiO2 deposition yield versus aluminon concentration.125 The results of the thermogravimetric analysis showed co-deposition of TiO2 and ATH. The amount of ATH in the composites can be varied by the variation of aluminon concentration in the suspensions.125
Another important finding was the possibility of efficient dispersion and EPD of MWCNT and graphene using aluminon and PANa. The EPD method allowed controlled EPD of MWCNT and graphene films on conductive substrates.146 Further development of this method allowed the fabrication of composite TiO2–MWCNT films using aluminon as a co-dispersant for the individual components.146
6. Future outlook
This section describes new trends in EPD of nanostructured films and composites and combined electrochemical methods based on EPD and other electrochemical techniques.As illustrated in this review, new dispersing agents offer advantage of strong adsorption on inorganic particles and can be used for the EPD of various materials. Therefore, further development of dispersants form catechol, salicylic acid, gallic acid and chromotropic acid families is emerging as a new area of technological and scientific interest. Significant opportunities lie in the application of new dispersing agents for agglomerate free synthesis of oxide nanoparticles of controlled size. Especially interesting is the possibility of modification of magnetic, optical, photovoltaic and other properties of oxide nanoparticles by the adsorbed dispersant molecules. Dopamine has recently been investigated for dispersion and modification of properties of quantum dots.148 The discovery of dispersing agents with film forming properties opens the door to making advanced films and composites. Moreover, new EPD strategies pave the way to the template free synthesis of fibers. The π–π stacking of PAH molecules (Fig. 15) resulted in anisotropic particle growth during EPD. It is important to note that fibers of other anionic and cationic dyes from pyrene147 and crystal violet149 families were recently prepared by EPD method. Therefore, EPD represents an alternative to the chemical self-assembly methods,150–154 which allowed the fabrication of fibers, nanotubes, nanowires, and nanorods from solutions of polyaromatic molecules. Such materials exhibited advanced and tunable photovoltaic, optical, and fluorescence properties. It was found that EPD method can be used for the fabrication of similar materials. Moreover, EPD allowed the formation of films, containing oriented rod-like particles.149 The π–π stacking of crystal violet molecules resulted in anisotropic particle growth during EPD.149
New dispersing agents have created interesting opportunities in the development of composites. Feasibility studies showed that various molecules can be used as co-dispersants for different oxides, carbon nanotubes and graphene. The possibility of efficient dispersion of pristine carbon nanotubes in suspensions and in composite films is of critical importance for many applications of carbon nanotubes, based on their conductive and mechanical properties. The new strategy can be used for the deposition of multilayers and functionally graded materials, containing oxide particles, carbon nanotubes, graphene and nanofibers of organic dyes.
New dispersing agents offer great promise for the fabrication of advanced composites using combined electrochemical methods, based on EPD of oxides and electroplating of metals or electropolymerization of polymers. Cationic CB is of special interest for the dispersion of ceramic particles and fabrication of composite metal–ceramic films by combining EPD of the ceramic particles and cathodic electroplating of metals. Tiron,155 CHR,156 AR,157 SA,158,159 SSA160 and PV135 are under intensive investigations for applications as anionic dopants for electropolymerization of polypyrrole films. Such films showed improved conductivity and enhanced electrochemical performance. Moreover, adherent films were obtained on non-noble substrates due to surface complexation of metal atoms on the substrate surface. It was demonstrated that tiron,155 CHR,156 AR157 and other molecules from catechol, salicylic acid and chromotropic acid families reduced electropolymerization potential of pyrrole. As a result the problem related to anodic dissolution of non-noble substrates during anodic electropolymerization of pyrrole was diminished. The strong adsorption of the molecules on the substrates facilitated charge transfer during electropolymerization. This approach opens the door to many applications of polypyrrole films in energy storage devices and protective coatings. It was found that electrochemical polymerization of polypyrrole can be combined with EPD of other functional materials using tiron161 and PV135 as anionic dopants for polypyrrole and charged dispersants for other materials. It is expected that this method will allow the fabrication of various composites containing nanoparticles of various functional materials in a conductive polymer matrix. The modification of polypyrrole with dopamine is a promising technique, which allowed improved polymer adhesion.162
New dispersion agents can be used for nanoparticle synthesis, surface modification and EPD of advanced films for application in electrochemical supercapacitors, batteries, photovoltaic, magnetic devices, catalysts and sensors, protective and flame retardant coatings. Preliminary work has shown that efficient dispersion strategies can be used for the synthesis of nanoparticles of controlled shape and size, fabrication of coated ceramic particles and carbon nanotubes. Important advantages of the new dispersing agents can be utilized for the fabrication of various materials and devices by other colloidal techniques.
7. Conclusions
Anionic and cationic molecules from catechol, salicylic acid, gallic acid and chromotropic acid families are promising charging and dispersing agents for EPD of oxide films. The molecules showed good adsorption on oxide particles due to interactions of their adjacent phenolic OH groups and COOH groups with metal atoms on the particle surface and allowed the fabrication of stable suspensions. New dispersing agents are well suited for co-deposition of various oxide materials. The adsorption of new dispersing agents imparted important functional properties to oxide nanoparticles and allowed synthesis of non-agglomerated nanoparticles of controlled size. The EPD technique can be used for the fabrication of organic fibers with important functional properties by a template free method. Charged organic dyes from catechol and salicylate families are attractive dispersants for co-deposition of oxides, carbon nanotubes, graphene and other materials. New dispersing agents can be used as multifunctional additives for the fabrication of composite coatings. Particularly important for future applications are dispersing agents with film forming properties. The growing colloidal and interface science of advanced dispersants opens the door to many advanced applications of EPD for the fabrication of functional materials for electrochemical energy storage, photovoltaic and biomedical devices, protective and flame retardant coatings, sensors and catalysis.Acknowledgements
The authors gratefully acknowledge the financial support of the Natural Sciences and Engineering Research Council of Canada.References
- I. Zhitomirsky, Adv. Colloid Interface Sci., 2002, 97, 277 CrossRef
.
- E. A. Olevsky, X. Wang, A. Maximenko and M. A. Meyers, J. Am. Ceram. Soc., 2007, 90, 3047 CrossRef CAS PubMed
- G. Cao, J. Phys. Chem. B, 2004, 108, 19921 CrossRef CAS
- S. V. Mahajan, J. Cho, M. S. P. Shaffer, A. R. Boccaccini and J. H. Dickerson, J. Eur. Ceram. Soc., 2010, 30, 1145 CrossRef CAS PubMed
- J. B. Talbot, E. Sluzky and S. K. Kurinec, J. Mater. Sci., 2004, 39, 771 CrossRef CAS
- B. Ferrari and R. Moreno, J. Eur. Ceram. Soc., 2010, 30, 1069 CrossRef CAS PubMed
- A. R. Boccaccini, J. Cho, J. A. Roether, B. J. C. Thomas, E. Jane Minay and M. S. P. Shaffer, Carbon, 2006, 44, 3149 CrossRef CAS PubMed
- L. Besra and M. Liu, Prog. Mater. Sci., 2007, 52, 1 CrossRef CAS PubMed
- O. O. Van der Biest and L. J. Vandeperre, Annu. Rev. Mater. Sci., 1999, 29, 327 CrossRef CAS
- P. Sarkar and P. S. Nicholson, J. Am. Ceram. Soc., 1996, 79, 1987 CrossRef CAS PubMed
- A. R. Boccaccini, C. Kaya and K. K. Chawla, Composites, Part A, 2001, 32, 997 CrossRef
- A. R. Boccaccini, J. A. Roether, B. J. C. Thomas, M. S. P. Shaffer, E. Chavez, E. Stoll and E. Jane Minay, J. Ceram. Soc. Jpn., 2006, 114, 1 CrossRef CAS
- B. Ferrari and R. Moreno, J. Eur. Ceram. Soc., 1997, 17, 549 CrossRef CAS
- B. Ferrari and R. Moreno, J. Electrochem. Soc., 2000, 147, 2987 CrossRef CAS PubMed
- B. Ferrari, R. Moreno, L. Hernan, M. Melero, J. Morales and A. Caballero, J. Eur. Ceram. Soc., 2007, 27, 3823 CrossRef CAS PubMed
- P. Mondragon-Cortez and G. Vargas-Gutierrez, Adv. Eng. Mater., 2003, 5, 812 CrossRef CAS
- P. Mondragon-Cortez and G. Vargas-Gutierrez, Mater. Lett., 2004, 58, 1336 CrossRef CAS PubMed
- D. Hanaor, M. Michelazzi, C. Leonelli and C. C. Sorrell, J. Eur. Ceram. Soc., 2012, 32, 235 CrossRef CAS PubMed
- A. J. Krejci, I. Gonzalo-Juan and J. H. Dickerson, ACS Appl. Mater. Interfaces, 2011, 3, 3611 CAS
- S. A. Hasan, J. L. Rigueur, R. R. Harl, A. J. Krejci, I. Gonzalo-Juan, B. R. Rogers and J. H. Dickerson, ACS Nano, 2010, 4, 7367 CrossRef CAS PubMed
- H. Mazor, D. Golodnitsky, L. Burstein, A. Gladkich and E. Peled, J. Power Sources, 2012, 198, 264 CrossRef CAS PubMed
- S. A. Hasan, D. W. Kavich, S. V. Mahajan and J. H. Dickerson, Thin Solid Films, 2009, 517, 2665 CrossRef CAS PubMed
- C. Kaya, Ceram. Int., 2008, 34, 1843 CrossRef CAS PubMed
- L.-Q. Wu, A. P. Gadre, H. Yi, M. J. Kastantin, G. W. Rubloff, W. E. Bentley, G. F. Payne and R. Ghodssi, Langmuir, 2002, 18, 8620 CrossRef CAS
- S. L. Quale and J. B. Talbot, J. Electrochem. Soc., 2007, 154, K25 CrossRef CAS PubMed
- B. P. Lee, P. B. Messersmith, J. N. Israelachvili and J. H. Waite, Annu. Rev. Mater. Res., 2011, 41, 99 CrossRef CAS PubMed
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426 CrossRef CAS PubMed
- H. Lee, B. P. Lee and P. B. Messersmith, Nature, 2007, 448, 338 CrossRef CAS PubMed
- L. Petrone, Adv. Colloid Interface Sci., 2013, 195–196, 1 CrossRef CAS PubMed
- J. P. Cornard and C. Lapouge, J. Phys. Chem. A, 2004, 108, 4470 CrossRef CAS
- I. A. Jankovic, Z. V. Saponjic, M. I. Comor and J. M. Nedeljkovic, J. Phys. Chem. C, 2009, 113, 12645 CAS
- M. Yu and T. J. Deming, Macromolecules, 1998, 31, 4739 CrossRef CAS PubMed
- Q. Ye, F. Zhou and W. Liu, Chem. Soc. Rev., 2011, 40, 4244 RSC
- J. L. Dalsin, B.-H. Hu, B. P. Lee and P. B. Messersmith, J. Am. Chem. Soc., 2003, 125, 4253 CrossRef CAS PubMed
- K. V. S. Devi, B. R. Raju and G. N. Rao, Acta Chim. Slov., 2010, 57, 398 CAS
- C. Fant, J. Hedlund, F. Höök, M. Berglin, E. Fridell and H. Elwing, J. Adhes., 2010, 86, 25 CrossRef CAS
- S. Bahri, C. M. Jonsson, C. L. Jonsson, D. Azzolini, D. A. Sverjensky and R. M. Hazen, Environ. Sci. Technol., 2011, 45, 3959 CrossRef CAS PubMed
- H. Lee, N. F. Scherer and P. B. Messersmith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12999 CrossRef CAS PubMed
- S. Fireman-Shoresh, I. Turyan, D. Mandler, D. Avnir and S. Marx, Langmuir, 2005, 21, 7842 CrossRef CAS PubMed
- X. Fan, L. Lin, J. L. Dalsin and P. B. Messersmith, J. Am. Chem. Soc., 2005, 127, 15843 CrossRef CAS PubMed
- J. Sedó, J. Saiz-Poseu, F. Busqué and D. Ruiz-Molina, Adv. Mater., 2013, 25, 653 CrossRef PubMed
- J. H. Waite, Nat. Mater., 2008, 7, 8 CrossRef CAS PubMed
- M. Maue and T. Schrader, Angew. Chem., Int. Ed., 2005, 44, 2265 CrossRef CAS PubMed
- H.-J. Koh, M. F. Hirshman, H. He, Y. Li, Y. Manabe, J. A. Balschi and L. J. Goodyear, Biochem. J., 2007, 403, 473 CrossRef CAS PubMed
- S. Kim and C. B. Park, Biomaterials, 2010, 31, 6628 CrossRef CAS PubMed
- D. K. Nagesha, B. D. Plouffe, M. Phan, L. H. Lewis, S. Sridhar and S. K. Murthy, J. Appl. Phys., 2009, 105, 07B317 CrossRef PubMed
- K. V. Korpany, F. Habib, M. Murugesu and A. S. Blum, Mater. Chem. Phys., 2013, 138, 29 CrossRef CAS PubMed
- G.-L. Wang, J.-J. Xu and H.-Y. Chen, Biosens. Bioelectron., 2009, 24, 2494 CrossRef CAS PubMed
- S. Varaganti and G. Ramakrishna, J. Phys. Chem. C, 2010, 114, 13917 CAS
- T. Rajh, L. X. Chen, K. Lukas, T. Liu, M. C. Thurnauer and D. M. Tiede, J. Phys. Chem. B, 2002, 106, 10543 CrossRef CAS
- B. M. Rabatic, N. M. Dimitrijevic, R. E. Cook, Z. V. Saponjic and T. Rajh, Adv. Mater., 2006, 18, 1033 CrossRef CAS
- A. S. Goldmann, C. Schödel, A. Walther, J. Yuan, K. Loos and A. H. Müller, Macromol. Rapid Commun., 2010, 31, 1608 CrossRef CAS PubMed
- K. Wu and I. Zhitomirsky, Int. J. Appl. Ceram. Technol., 2011, 8, 920 CrossRef CAS PubMed
- W. Huang, P. Jiang, C. Wei, D. Zhuang and J. Shi, J. Mater. Res., 2008, 23, 1946 CrossRef CAS
- D. Gutiérrez-Tauste, X. Domènech, C. Domingo and J. A. Ayllón, Thin Solid Films, 2008, 516, 3831 CrossRef PubMed
- L. De la Garza, Z. V. Saponjic, N. M. Dimitrijevic, M. C. Thurnauer and T. Rajh, J. Phys. Chem. B, 2006, 110, 680 CrossRef CAS PubMed
- A. Hayat, D. Andreescu, G. Bulbul and S. Andreescu, J. Colloid Interface Sci., 2014, 418, 240 CrossRef CAS PubMed
- Y. Wang and I. Zhitomirsky, Langmuir, 2009, 25, 9684 CrossRef CAS PubMed
- K. Wu, P. Imin, A. Adronov and I. Zhitomirsky, Mater. Chem. Phys., 2011, 125, 210 CrossRef CAS PubMed
- Á. Valero-Navarro, M. Gómez-Romero, J. F. Fernández-Sánchez, P. A. Cormack, A. Segura-Carretero and A. Fernández-Gutiérrez, J. Chromatogr., A, 2011, 1218, 7289 CrossRef PubMed
- F. S. De Souza and A. Spinelli, Corros. Sci., 2009, 51, 642 CrossRef CAS PubMed
- W. J. Barreto, R. A. Ando, B. M. Estevao and K. P. D. S. Zanoni, Spectrochim. Acta, Part A, 2012, 92, 16 CrossRef CAS PubMed
- Y. Sun and I. Zhitomirsky, Mater. Lett., 2012, 73, 190 CrossRef CAS PubMed
- V. K. Pogorelyi, O. A. Kazakova, V. N. Barvinchenko, O. V. Smirnova, E. M. Pakhlov and V. M. Gun'Ko, Colloid J., 2007, 69, 203 CrossRef CAS
- O. A. Dovbii, O. A. Kazakova and N. A. Lipkovskaya, Colloid J., 2006, 68, 707 CrossRef CAS
- Y. Wang and I. Zhitomirsky, J. Colloid Interface Sci., 2012, 380, 8 CrossRef CAS PubMed
- Q. Chen, Y. Jia, S. Liu, G. Mogilevsky, A. Kleinhammes and Y. Wu, J. Phys. Chem. C, 2008, 112, 17331 CAS
- H. Gulley-Stahl, P. A. Hogan, W. L. Schmidt, S. J. Wall, A. Buhrlage and H. A. Bullen, Environ. Sci. Technol., 2010, 44, 4116 CrossRef CAS PubMed
- D. Vasudevan and A. T. Stone, Environ. Sci. Technol., 1996, 30, 1604 CrossRef CAS
- D. Vasudevan and A. T. Stone, J. Colloid Interface Sci., 1998, 202, 1 CrossRef CAS
- Y. S. Hwang and J. J. Lenhart, J. Colloid Interface Sci., 2009, 336, 200 CrossRef CAS PubMed
- O. Klug and W. Forsling, Langmuir, 1999, 15, 6961 CrossRef CAS
- S.-C. Li, J.-G. Wang, P. Jacobson, X. Q. Gong, A. Selloni and U. Diebold, J. Am. Chem. Soc., 2009, 131, 980 CrossRef CAS PubMed
- S. Tunesi and M. A. Anderson, Langmuir, 1992, 8, 487 CrossRef CAS
- K. D. Dobson and A. J. McQuillan, Spectrochim. Acta, Part A, 2000, 56, 557 CrossRef CAS
- L. Boilet, J. P. Cornard and C. Lapouge, J. Phys. Chem. A, 2005, 109, 1952 CrossRef CAS PubMed
- M. V. Biber and W. Stumm, Environ. Sci. Technol., 1994, 28, 763 CrossRef CAS PubMed
- M. S. Ata and I. Zhitomirsky, Adv. Appl. Ceram., 2012, 111, 345 CrossRef CAS PubMed
- J. Peyre, V. Humblot, C. Méthivier, J.-M. Berjeaud and C.-M. Pradier, J. Phys. Chem. B, 2012, 116, 13839 CrossRef CAS PubMed
- T. Togashi, S. Takami, K. Kawakami, H. Yamamoto, T. Naka, K. Sato, K. Abe and T. Adschiri, J. Mater. Chem., 2012, 22, 9041 RSC
- L. Jiang, L. Gao and Y. Liu, Colloids Surf., A, 2002, 211, 165 CrossRef CAS
- R. Laucournet, C. Pagnoux, T. Chartier and J. F. Baumard, J. Am. Ceram. Soc., 2000, 83, 2661 CrossRef CAS PubMed
- L. Jiang and L. Gao, Mater. Chem. Phys., 2003, 80, 157 CrossRef CAS
- A. Papo and L. Piani, Part. Sci. Technol., 2007, 25, 375 CrossRef CAS
- S. Lebrette, C. Pagnoux and P. Abelard, J. Eur. Ceram. Soc., 2006, 26, 2727 CrossRef CAS PubMed
- S. Lebrette, C. Pagnoux and P. Abélard, J. Colloid Interface Sci., 2004, 280, 400 CrossRef CAS PubMed
- D. Schiemann, P. Alphonse and P.-L. Taberna, J. Mater. Res., 2013, 28, 2023 CrossRef CAS
- M. Guedes, J. M. F. Ferreira and A. C. Ferro, J. Colloid Interface Sci., 2009, 330, 119 CrossRef CAS PubMed
- C. Kawaguti, C. Santilli and S. Pulcinelli, J. Non-Cryst. Solids, 2008, 354, 4790 CrossRef CAS PubMed
- S. Belin, L. R. B. Santos, V. Briois, A. Lusvardi, C. V. Santilli, S. H. Pulcinelli, T. Chartier and A. Larbot, Colloids Surf., A, 2003, 216, 195 CrossRef CAS
- T. Sugimoto, H. Itoh and T. Mochida, J. Colloid Interface Sci., 1998, 205, 42 CrossRef CAS PubMed
- T. Navizi, E. Salahi, M. Ghafari and I. Mobasherpour, Ceram. Int., 2010, 36, 1945 CrossRef CAS PubMed
- S. Moghadas, A. Maghsoudipour, M. Alizadeh and T. Ebadzadeh, Ceram. Int., 2011, 37, 2015 CrossRef CAS PubMed
- B. J. Briscoe, A. U. Khan and P. F. Luckham, J. Eur. Ceram. Soc., 1998, 18, 2169 CrossRef CAS
- S. Mei, J. Yang and J. M. F. Ferreira, Ceram. Int., 2003, 29, 785 CrossRef CAS
- A.-L. Penard, F. Rossignol, H. Nagaraja, C. Pagnoux and T. Chartier, J. Eur. Ceram. Soc., 2005, 25, 1109 CrossRef CAS PubMed
- M. Hori, C. Pagnoux, J. F. Baumard and M. Nogami, J. Mater. Sci., 2007, 42, 80 CrossRef CAS PubMed
- P. Z. Araujo, P. J. Morando and M. A. Blesa, Langmuir, 2005, 21, 3470 CrossRef CAS PubMed
- Y. Wang and I. Zhitomirsky, Colloids Surf., A, 2010, 369, 211 CrossRef CAS PubMed
- A. R. Studart, E. Amstad and L. J. Gauckler, Langmuir, 2007, 23, 1081 CrossRef CAS PubMed
- E. Tombacz, I. Y. Toth, D. Nesztor, E. Illes, A. Hajdu, M. Szekeres and L. Vekas, Colloids Surf., A, 2013, 435, 91 CrossRef CAS PubMed
- K. Wu, Y. Wang and I. Zhitomirsky, J. Colloid Interface Sci., 2010, 352, 371 CrossRef CAS PubMed
- L. Zhao, J. Hu and B. He, Adv. Mater. Res., 2011, 335–336, 1262 CrossRef CAS
- X. Zhang, Y. Hou, P. Hu and C. Hong, J. Eur. Ceram. Soc., 2012, 32, 3463 CrossRef CAS PubMed
- W. Wang, Q. Chen, C. Jiang, D. Yang, X. Liu and S. Xu, Colloids Surf., A, 2007, 301, 73 CrossRef CAS PubMed
- K. Kratochvilová, I. Hoskovcová, J. Jirkovský, J. Klíma and J. Ludvík, Electrochim. Acta, 1995, 40, 2603 CrossRef
- J. Moser, S. Punchihewa, P. P. Infelta and M. Graetzel, Langmuir, 1991, 7, 3012 CrossRef CAS
- N. Kallay, T. Preocanin, J. Markovic and D. Kovacevic, Colloids Surf., A, 2007, 306, 40 CrossRef CAS PubMed
- H. Husin, Y.-K. Leong and J. Liu, Colloids Surf., A, 2012, 402, 159 CrossRef CAS PubMed
- M. S. Ata and I. Zhitomirsky, Mater. Manuf. Processes, 2013, 28, 1014 CAS
- X. Xie, Y. Wu, Y. Kong, Z. Zhang and X. Zhou, Colloids Surf., A, 2012, 408, 104 CrossRef CAS PubMed
- W. Yin, X. Chen, M. Cao, C. Hu and B. Wei, J. Phys. Chem. C, 2009, 113, 15897 CAS
- S. Li, F. Zheng, X. Liu, F. Wu, N. Deng and J. Yang, Chemosphere, 2005, 61, 589 CrossRef CAS PubMed
- Y. Sun, Y. Wang and I. Zhitomirsky, Colloids Surf., A, 2013, 418, 131 CrossRef CAS PubMed
- A. Begerhotta, H. C. Tandon and P. B. Mathur, J. Power Sources, 1988, 24, 341 CrossRef CAS
- B. Pandit and U. Chudasama, Colloids Surf., A, 1998, 132, 145 CrossRef CAS
- E. Destandau, V. Alain and E. Bardez, Anal. Bioanal. Chem., 2004, 378, 402 CrossRef CAS PubMed
- M. S. Masoud, E. A. Khalil, A. M. Hindawy, A. E. Ali and E. F. Mohamed, Spectrochim. Acta, Part A, 2004, 60, 2807 CrossRef PubMed
- A. Noorbakhsh and A. Salimi, Electrochim. Acta, 2009, 54, 6312 CrossRef CAS PubMed
- S. Yadav and C. Lal, Energy Convers. Manage., 2013, 66, 271 CrossRef CAS PubMed
- M. Du, B. Guo and D. Jia, Polym. Int., 2010, 59, 574 CAS
- M. Du, B. Guo and D. Jia, Eur. Polym. J., 2006, 42, 1362 CrossRef CAS PubMed
- M. Yang, D. Li, T. Zhao and J. Ma, J. Dispersion Sci. Technol., 2010, 31, 563 CrossRef CAS
- P. M. Jayaweera and T. A. U. Jayarathne, Surf. Sci., 2006, 600, L297 CrossRef CAS PubMed
- Y. Sun, M. S. Ata and I. Zhitomirsky, J. Colloid Interface Sci., 2012, 369, 395 CrossRef CAS PubMed
- F. Fu, Z. Gao, L. Gao and D. Li, Ind. Eng. Chem. Res., 2011, 50, 9712 CrossRef CAS
- T. Polubesova, M. Epstein, S. Yariv, I. Lapides and S. Nir, Appl. Clay Sci., 2004, 24, 177 CrossRef CAS PubMed
- T. Moriguchi, K. Yano, S. Nakagawa and F. Kaji, J. Colloid Interface Sci., 2003, 260, 19 CrossRef CAS
- G. Ramakrishna, H. N. Ghosh, A. K. Singh, D. K. Palit and J. P. Mittal, J. Phys. Chem. B, 2001, 105, 12786 CrossRef CAS
- S. Deki, Y. Kodama and M. Mizuhata, J. Ceram. Soc. Jpn., 2009, 117, 326 CrossRef CAS
- Y. Wang, Y. Liu and I. Zhitomirsky, J. Mater. Chem. A, 2013, 1, 12519 CAS
- M. S. Ata, G. Z. Zhu, G. A. Botton and I. Zhitomirsky, Adv. Appl. Ceram., 2014, 113, 22 CrossRef CAS PubMed
- H. Chang, H.-T. Su, W.-A. Chen, K. David Huang, S.-H. Chien, S.-L. Chen and C.-C. Chen, Sol. Energy, 2010, 84, 130 CrossRef CAS PubMed
- W. Jarernboon, S. Pimanpang, S. Maensiri, E. Swatsitang and V. Amornkitbamrung, Thin Solid Films, 2009, 517, 4663 CrossRef CAS PubMed
- X. Li and I. Zhitomirsky, J. Power Sources, 2013, 221, 49 CrossRef CAS PubMed
- A. R. Boccaccini, J. Cho, T. Subhani, C. Kaya and F. Kaya, J. Eur. Ceram. Soc., 2010, 30, 1115 CrossRef CAS PubMed
- H. Atabey and H. Sari, J. Chem. Eng. Data, 2011, 56, 3866 CrossRef CAS
- A. Janowski, J. Rzeszotarska and N. Sadlej, J. Lumin., 1972, 5, 453 CrossRef CAS
- Y. Su and I. Zhitomirsky, J. Phys. Chem. B, 2013, 117, 1563 CrossRef CAS PubMed
- G. S. Baghel and C. P. Rao, Polyhedron, 2009, 28, 3507 CrossRef CAS PubMed
- E. Casassas and A. Izquierdo-Ridorsa, Polyhedron, 1990, 9, 1191 CrossRef
- K. Shqau, M. L. Mottern, D. Yu and H. Verweij, J. Am. Ceram. Soc., 2006, 89, 1790 CrossRef CAS PubMed
- B. J. Briscoe, A. U. Khan and P. F. Luckham, J. Eur. Ceram. Soc., 1998, 18, 2141 CrossRef CAS
- A. U. Khan, N. Mahmood and P. F. Luckham, J. Dispersion Sci. Technol., 2012, 33, 1210 CrossRef CAS
- C. Falamaki, Z. Khakpour and A. Aghaie, J. Membr. Sci., 2005, 263, 103 CrossRef CAS PubMed
- M. S. Ata, Y. Sun, X. Li and I. Zhitomirsky, Colloids Surf., A, 2012, 398, 9 CrossRef CAS PubMed
- Y. Su and I. Zhitomirsky, J. Colloid Interface Sci., 2013, 392, 247 CrossRef CAS PubMed
- M. Bloemen, D. Debruyne, P.-J. Demeyer, K. Clays, A. Gils, N. Geukens, C. Bartic and T. Verbiest, RSC Adv., 2014, 4, 10208 RSC
- Y. Su and I. Zhitomirsky, J. Colloid Interface Sci., 2013, 399, 46 CrossRef CAS PubMed
- Y. Kamikawa and T. Kato, Langmuir, 2007, 23, 274 CrossRef CAS PubMed
- J. Xiao, Y. Liu, Y. Li, J. Ye, Y. Li, X. Xu, X. Li, H. Liu, C. Huang, S. Cui and D. Zhu, Carbon, 2006, 44, 2785 CrossRef CAS PubMed
- Y. Chen, B. Zhu, Y. Han and Z. Bo, J. Mater. Chem., 2012, 22, 4927 RSC
- X. Zhang, X. Zhang, W. Shi, X. Meng, C. Lee and S. Lee, J. Phys. Chem. B, 2005, 109, 18777 CrossRef CAS PubMed
- Z. Wang, M. Liu, Y. Xie and C. Gao, J. Mater. Chem., 2012, 22, 2855 RSC
- D. E. Tallman, C. Vang, G. G. Wallace and G. P. Bierwagen, J. Electrochem. Soc., 2002, 149, C173 CrossRef CAS PubMed
- D. K. Ariyanayagamkumarappa and I. Zhitomirsky, Synth. Met., 2012, 162, 868 CrossRef CAS PubMed
- S. Chen and I. Zhitomirsky, Mater. Lett., 2013, 98, 67 CrossRef CAS PubMed
- E. Hermelin, J. Petitjean, S. Aeiyach, J. C. Lacroix and P. C. Lacaze, J. Appl. Electrochem., 2001, 31, 905 CrossRef CAS
- V. K. Gade, D. J. Shirale, P. D. Gaikwad, P. A. Savale, K. P. Kakde, H. J. Kharat and M. D. Shirsat, Int. J. Polym. Mater., 2007, 56, 167 CrossRef CAS
- X. Li, P. Imin, A. Adronov and I. Zhitomirsky, Mater. Lett., 2012, 68, 24 CrossRef CAS PubMed
- C. Shi and I. Zhitomirsky, Surf. Eng., 2011, 27, 655 CrossRef CAS PubMed
- W. Zhang, F. K. Yang, Z. Pan, J. Zhang and B. Zhao, Macromol. Rapid Commun., 2014, 35, 350 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2014 |