DOI:
10.1039/C4RA02176B
(Paper)
RSC Adv., 2014,
4, 28928-28936
Tuning the surface microstructure and gradient properties of polymers with photopolymerizable polysiloxane-modified nanogels†
Received
12th March 2014
, Accepted 29th April 2014
First published on 29th April 2014
Abstract
This paper reports a series of photopolymerizable polysiloxane-modified nanogels for regulating the surface microstructure and gradient properties of polymers, which were synthesized by solution polymerization with different feed ratios of methacrylate-modified polysiloxane, urethane dimethacrylate (UDMA) and isobornyl methacrylate (IBMA) in the presence of a thiol chain transfer agent. The nanogel structures and compositions were characterized by proton nuclear magnetic resonance (1H-NMR), Fourier transform-infrared spectroscopy (FT-IR), transmission electron microscopy (TEM), gel permeation chromatography (GPC) and differential scanning calorimetry (DSC). The dispersion of these nanogels in triethylene glycol dimethacrylate (TEGDMA) can delay the onset and reduce the magnitude of shrinkage stress during polymerization without compromising the mechanical properties of the resulting polymers. Most importantly, as demonstrated by elemental analysis and X-ray photoelectron spectroscopy (XPS), the nanogels exhibit good self-floating ability in the monomer–polymer matrix and the increase in polysiloxane content in the nanogel can enhance the self-floating capability due to the lower surface tension and energy associated with the polysiloxane component. As a result, the polysiloxane-modified nanogels can spontaneously form a concentration gradient that can be locked in upon photopolymerization, leading to a well-controlled heterogeneous polymer that presents a gradient change in thermal stability. With the increase in polysiloxane content, the thermal stability of the polymer was improved significantly. Furthermore, the enrichment of the nanogel on the surface resulting from the good self-floating ability can reduce the dispersion surface energy of the gradient polymer film and generate a more hydrophobic surface with an altered surface microstructure. These photopolymerizable polysiloxane-modified nanogels are demonstrated to have potentially broad applications in the preparation of gradient polymers with controlled surface properties.
Introduction
Polymeric gradient materials (PGMs) have gained considerable interest in recent years due to the continuous composition or microstructural variability in the polymers.1,2 As highly versatile functional materials, PGMs are able to achieve certain properties that cannot be fulfilled by homogeneous materials, for example, the mechanical properties of PGMs continuously vary in space,3 and have been widely used in aerospace, energy, electronics, medicine, optics and other industries.4,5 Regulation of the gradient properties and microstructures of polymers is of vital importance in preparing an ideal gradient polymer. Currently, a series of methods6,7 are used to regulate the gradient properties of polymers, including changing the light intensity,8 applying a gradient temperature field,9 varying the gradient solution concentration10 and utilizing magnetic separation.11,12 However, these methods involve multiple steps and the use of large amounts of solvents thereby increasing cost and pollution, and decreasing convenience. Hence, efforts to develop simpler, more environmentally friendly and convenient methods to regulate gradient properties remains a challenging goal.
Nanogels, which are internally crosslinked and cyclized single or multi-chain polymeric particles with three-dimensional polymer networks,13 have great potential for use in drug delivery,14 dental materials,15 tissue engineering,16 photonic materials,17 modifiers for coatings and polymer composites18 because of their tunable chemical compositions and three-dimensional physical structures, large number of chain ends, good mechanical properties, and biocompatibility. It has been demonstrated that high molecular weight, reactive nanogels of approximately 10 nm in dimension, when dispersed in and swollen by matrix monomers, can be used to reduce polymerization shrinkage and stress, without compromising other critical polymer properties,19 and help to reinforce the mechanical properties of the final polymer network by physical entanglement and potential covalent crosslinking between the nanogel and the resin matrix.
Polysiloxanes have many unique properties20 such as good resistance to high temperatures, excellent weatherability, low surface tension and energy, a low dielectric constant and high resistance to an array of chemical substances due to their unusual molecular structures. Specifically, the particularly low surface tension of polysiloxane provides the potential for a self-floating ability that results in the spontaneous formation of a concentration gradient distribution and enrichment at the surface, which is very valuable for the control of gradient properties, surface microstructure and the properties of gradient materials.21,22 This stimulated our interest to introduce polysiloxane into nanogels to engender some new desirable properties, as well as to examine the possibility of preparing functionally and structurally gradated polymers with controlled surface properties. Obviously, the polysiloxane content in the nanogel is a vital factor for control of the dispersion and character of these nanogels within secondary polymer networks.
In the current study, a series of nanogel compositions containing different polysiloxane contents were synthesized with different ratios of a methacrylate-modified polysiloxane, urethane dimethacrylate (UDMA) and isobornyl methacrylate (IBMA). Formulations were prepared by dispersing the nanogels into a dimethacrylate monomer and a variety of properties were evaluated, including rheology, photopolymerization kinetics, shrinkage stress and glass transition temperature of the cured samples at different nanogel loading concentrations. More importantly, the surface morphology and properties, elemental composition and thermostability of each nanogel-containing gradient polymer were investigated by scanning electron microscopy (SEM), energy-dispersive X-ray spectroscopy (EDS), contact angle goniometer (CA), X-ray photoelectron spectroscopy (XPS), and thermogravimetric analysis (TGA).
Experimental details
Materials and characterization
Isobornyl methacrylate (IBMA) and triethylene glycol dimethacrylate (TEGDMA) were donated by Eternal Chemical Co. Ltd. (Zhuhai, China). Urethane dimethacrylate (UDMA) and 2-isocyanatoethyl methacrylate (IEM) were purchased from Heowns Business License Co. Ltd. (Tianjin, China). Dual-end terminated methacrylate-modified polysiloxane (Mn = 380, methacrylate equivalent: 190 g mol−1) was donated by Shin-Etsu Chemical Co. Ltd. (Shanghai, China). Azobisisobutyronitrile (AIBN) was purchased from Xilong Chemical Co. Ltd. (Shantou, China). 2-Mercaptoethanol (ME) was supplied by Fuchen Chemical Reagents Factory (Tianjin, China). Dibutyltin dilaurate (DBTDL) was supplied by Shanghai Chemical Reagents Co. (Shanghai, China). 2,2-Dimethoxy-2-phenylacetophenone (DMPA) was obtained from Ciba Geigy Co. Ethyl acetate and dichloromethane (DCM) were purchased from Beijing Chemical Works (Beijing, China). All reagents were used as received without further purification. The structures of the main reagents used are shown in Fig. 1.
 |
| | Fig. 1 Structures of the monomers and reagents used in the study. | |
The FTIR spectra (scanned between 400 and 4000 cm−1) were obtained on a Nicolet 50XC spectrometer (Nicolet, USA). The 1H-NMR spectra were recorded on an AV400 unity spectrometer (Bruker, USA) operated at 400 MHz with CDCl3 as the solvent and tetramethylsilane as an internal standard. The molecular weight of the nanogel was determined by gel permeation chromatography using a Waters 515-2410 (Waters, USA) with tetrahydrofuran (1.0 mL min−1) used as the mobile phase. Gel permeation chromatography calibration was based on a series of linear polystyrene standards of known molecular weight and dispersity. The particle size and size distribution of the nanogel were determined by dynamic light scattering (DLS) using a Zetasizer NanoZS (ZEN 3600, Malvern, Germany) equipped with a 4 mW He–Ne solid-state laser operating at 633 nm. All measurements were performed three times in 0.01 w/v% aqueous solutions using disposable plastic cuvettes. Deionized water used to dilute each dispersion was ultrafiltered through a 0.20 μm membrane. Transmission electron microscopy (TEM) images were obtained with JEOL JEM-2010 high-resolution transmission electron microscopes. The accelerating voltage was 20.0 kV. Thermal stability was determined with a STA-449C simultaneous thermogravimetric analyzer (Netzsch) with the polymer samples run from 30 to 500 °C at a heating rate of 10 °C min−1. Differential scanning calorimetry (DSC) measurements were performed in nitrogen on 5–15 mg polymer samples at a heating rate of 20 °C min−1 over the temperature range of 25–200 °C using a Pyris 1 (Perkin Elmer). Dynamic mechanical thermal analyses (DMTAs) were performed on a DMTA-IV (Rheometric Scientific Co.) with samples which underwent post-cure conditioning at 160 °C overnight to fully cure, and were run from −50 to +250 °C at a heating rate of 5 °C min−1 with a frequency of 1 Hz. Elemental analysis for nitrogen was performed using a Vario EL cube (Elementar Analysensysteme, Germany) equipped with a thermal conductivity detector, operating over the temperature range of 950–1200 °C. The surface morphology of the polymer was observed using a scanning electron microscope (S-4700 Hitachi) with an accelerating voltage of 5.0 kV. The surface elements of the polymer samples were characterized using XPS (Thermo Electron Corporation, Escalab 250, Germany) and EDS (S-4700 Hitachi). Water contact angles on the surfaces of the polymer films were measured on a contact angle goniometer (OCA20, Data Physics Co., Germany).
Synthesis of polysiloxane-modified nanogels
Methacrylate-modified polysiloxane, UDMA and IBMA were added into a 500 mL three-necked round-bottom flask with a magnetic stirrer at molar ratios of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:70, 15:15:70 or 20:10:70, respectively. ME (7 mol% relative to monomers) was included as a chain transfer agent to aid in the prevention of macrogelation as well as to provide sites for post-polymerization refunctionalization with methacrylate groups. AIBN (1 wt% relative to monomers) was included as a free radical thermal initiator. A four-fold excess of ethyl acetate relative to the monomer (vol/mass) was introduced and the solution was heated in an oil bath at 90 °C for 2 h with a circulating water condenser in place.
:20:70, 15:15:70 or 20:10:70, respectively. ME (7 mol% relative to monomers) was included as a chain transfer agent to aid in the prevention of macrogelation as well as to provide sites for post-polymerization refunctionalization with methacrylate groups. AIBN (1 wt% relative to monomers) was included as a free radical thermal initiator. A four-fold excess of ethyl acetate relative to the monomer (vol/mass) was introduced and the solution was heated in an oil bath at 90 °C for 2 h with a circulating water condenser in place.
The resulting clear nanogel reaction mixture was added drop-wise to an eight-fold excess of hexane, which resulted in the precipitation of the polymeric materials. The precipitate was filtered and the residual solvent removed under reduced pressure to obtain the nanogel product as a dry powder in approximately 85% yield. 1H NMR characterization of the nanogels verified copolymer compositions similar to those of the monomer feed ratio used. In order to obtain reactive, photopolymerizable nanogels, the isolated nanogel was redispersed in 30 mL DCM in a sealed 50 mL round-bottom flask to which IEM (in the same molar concentration as ME) was added. The reaction mixture was stirred at room temperature in the presence of a catalytic amount of DBTDL. The reaction was monitored by periodic mid-FT-IR analysis until the isocyanate absorption peak had completely disappeared (Scheme 1). The resulting polysiloxane-modified nanogels, which were isolated by an analogous second precipitation step, are designated as Si10, Si15 and Si20, respectively, to represent the 10, 15 and 20 mol% components of polysiloxane in the nanogel. The IR spectra and 1H-NMR spectra of Si10, Si15 and Si20 display the following similar characteristics:
IR (KBr, cm−1): 3200–3500 cm−1 (–NH), 2925–2975 cm−1 (–CH3, –CH2), 1725 cm−1 (>C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1639 cm−1 (–CCH–), 1020–1092 cm−1 (Si–O–Si), 800–804 cm−1 (Si–CH3) (see Fig. S1†).
O), 1639 cm−1 (–CCH–), 1020–1092 cm−1 (Si–O–Si), 800–804 cm−1 (Si–CH3) (see Fig. S1†).
1H-NMR (CDCl3, ppm): δ5.6–6.2 (–CCH–), δ2.8–3.5 (–NH–), δ0.53–1.68 (–Si–CH2–), δ0.05–0.10 (–Si–CH3) (see Fig. S2†).
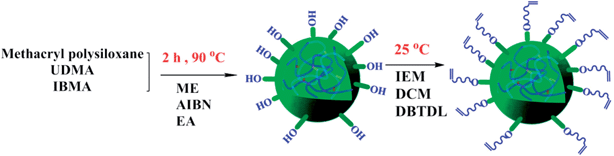 |
| | Scheme 1 Schematic representation of the synthesis route of the nanogel. | |
Mixture of resin with the nanogels
The polysiloxane-modified nanogel was added to TEGDMA in mass ratios of 10 wt% to 40 wt% in 10% increments. Unmodified TEGDMA was used as the control. DMPA (0.5 wt%, relative to resin) was added to each sample. Samples were mechanically agitated until the nanogel was fully dispersed to give optically clear monomeric solutions.
Synthesis of polymer cylinders containing the nanogel
The monomeric samples were placed in a vertical glass tube having an inner diameter of 6 mm to form a fluid column 8 cm in length that was then purged with nitrogen. After standing for 90 min, the fluid column was irradiated from the side by a high-pressure mercury lamp (incident light intensity = 5 mW cm−2, wavelength = 365 (±10) nm, recorded by UV radiometer (Photoelectric Instrument Factory, Beijing Normal University, Beijing, China)) with the tube fixed vertically on a turntable (speed ≈ 5 rpm) to ensure uniform irradiation. The silicon content and thermostability of different horizontal slices of the polymer (PTEGDMA) were measured by XPS and TGA.
Preparation of polymer film containing the nanogels in anaerobic conditions
A measured volume of the UV-resin formulations was dispensed onto a pre-cleaned glass slide and allowed to spread to a diameter of 20 mm to obtain a liquid resin film with a thickness of 0.3 mm. A cover glass was then placed over the film and ensured anaerobic conditions. After standing for 90 min, the deposited liquid film was exposed to a high-pressure mercury lamp (incident light intensity = 5 mW cm−2, recorded by UV radiometer) for 60 s to obtain the polymer films. The surface properties, microstructure, and glass transition temperature (Tg) of the films were measured by contact angle goniometry, scanning electron microscopy (SEM), energy-dispersive X-ray spectroscopy (EDS) and dynamic mechanical thermal analyses (DMTAs), respectively.
Rheology test
Viscosity measurements of the nanogel-modified TEGDMA resins were performed using an NDJ-79 viscometer (Shanghai Changji Instrument Co. Ltd, China). Defined volumes of the control and nanogel-modified resins were tested at 20 °C under the following conditions: 75 rpm and a run time of 30 s. Three replicates were conducted for each sample.
Simultaneous measurement of shrinkage stress and conversion
A cantilever beam-based tensometer (Paffenbarger Research Center, American Dental Association Foundation, Gaithersburg, MD) was utilized for the measurement of dynamic shrinkage stress. Instrument details and operation methods are provided in other publications.23 The dynamic stress development of each material (cylindrical shape of 6 mm diameter and 1.5 mm thickness, n = 3) was evaluated continuously during irradiation with 365 (±10) nm UV light at an intensity of 10 mW cm−2 for 15 min. During the real-time stress evaluation, transmission near-infrared spectroscopy was simultaneously incorporated to measure the reaction kinetics profile. The peak area of the first overtone absorbance of the methacrylate CH2 group at 6165 cm−1 was followed throughout the polymerization process. Three replicates were carried out for each material composition. The detailed experimental procedure describing these simultaneous characterization techniques has been reported previously.24
Flexural strength test
A universal testing machine (Mini-Bionix II, MTS, Eden Prairie, MN) was used to gather mechanical property data (at room temperature). Sample specimens (n = 3) with dimensions of 15 × 2 × 2 mm were photopolymerized under 365 (±10) nm UV light irradiation at 15 mW cm−2 intensity for 10 min. Flexural modulus and strength were determined in three-point bending on a 10 mm span at a crosshead speed of 1 mm min−1.
Nanogel self-floating ability
The nanogel was dispersed in dichloromethane to obtain homogeneous solutions with nanogel concentrations of 10 wt%. Each solution was placed in a vertical glass tube having an inner diameter of 3 cm and depth of 25 cm. After standing for different times (0 min, 10 min, 30 min, 60 min, 90 min, 120 min and 180 min), 1.0 mL aliquots were carefully removed from the top and bottom of the vertical glass tube. The concentration of nitrogen in each sample, which reflects the urethane functional group content of the nanogel, was detected by elemental analysis.
The polymer rod, as illustrated in ‘Synthesis of polymer cylinders containing the nanogel’, was cut into three even segments, and the elemental compositions and morphologies of the top, bottom and cut surfaces were observed by XPS, SEM and EDS to further investigate the nanogel self-floating ability (see Fig. 10 for details).
Results and discussion
Nanogel characterization
The GPC analyses, bulk Tg values and diameters of nanogels Si10, Si15 and Si20 are summarized in Table 1. With the increase in polysiloxane content, the weight average molecular weight (Mw) correspondingly decreased, possibly due to the smaller molecular weight of polysiloxane compared with UDMA. However, the polydispersity index (PDI) showed the opposite trend, because of normal distributions of chain numbers within the nanogels, intraparticle cyclization reactions and various interparticle reactions, but it is still relatively low for highly branched, multi-chain polymeric structures. Even with the flexible nature of polysiloxanes, it is interesting that the Tg values of the nanogels increased along with the polysiloxane concentration. The more than 10 °C increase in the bulk Tg values of the nanogels as the polysiloxane content was doubled may indicate that the remaining urethane functionality associated with the UDMA crosslinker is able to form stronger interactions because of the greater intraparticle mobility. Indeed, even though the nanogel molecular weight did not vary appreciably, the average diameter of the water-dispersed nanogel particles decreased from 59.6 nm for Si10 to 29.5 nm for Si20, which may be expected based on the high flexibility and hydrophobicity of polysiloxane. It is known that the Si–O bond is longer than the C–C bond, but the Si–O bond is so flexible that it is even able to pass through the planar 180°. The high flexibility might make molecular chain entanglement tighter, thereby decreasing the average diameters of the nanogels. The morphologies of the nanogels were probed by TEM (Fig. 2). TEM images show that the nanogels appear as microspheres and their average diameters are slightly smaller than those measured by DLS because of the transition of the assemblies from the swollen (DLS) to dried state (TEM).
Table 1 Mw, Tg and diameter characterization of the nanogels
| Nanogel |
Mw (Da) |
PDI |
Tg (°C) |
Diameter (nm) |
| DLS |
TEM |
| Si10 |
110000 |
2.11 |
97.1 |
59.6 |
52.9 |
| Si15 |
94800 |
2.92 |
99.3 |
39.0 |
32.1 |
| Si20 |
94700 |
3.05 |
109.4 |
29.5 |
23.4 |
 |
| | Fig. 2 Transmission electron micrographs of nanogels (a) Si10; (b) Si15 and (c) Si20. | |
Viscosity test
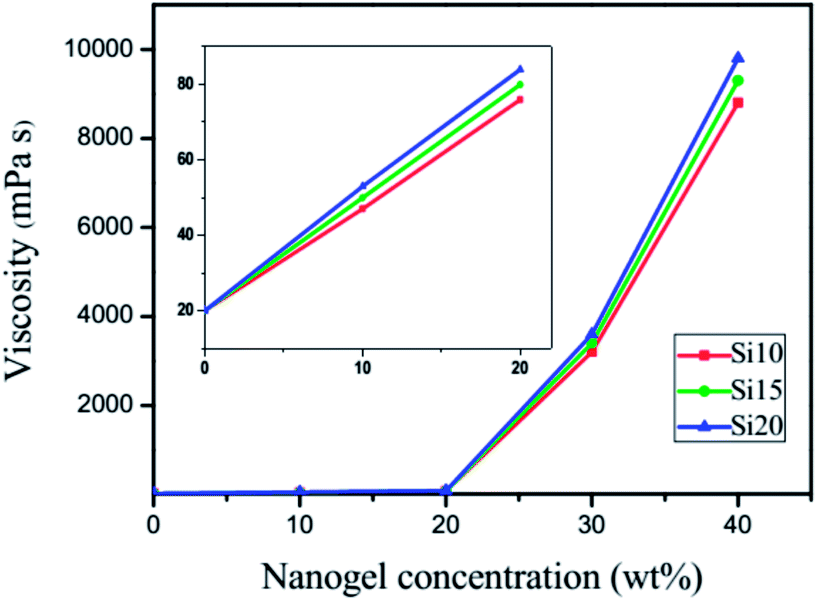
For a UV-curable resin, viscosity is an important parameter that directly affects substrate adaptability, the rate of polymerization and the properties of cured films. Viscosity measurements on uncured TEGDMA samples with various nanogel contents are shown in Fig. 3. With the addition of nanogels at different loading levels, the viscosity increased initially in a linear fashion followed by a logarithmic increase. The different nanogel materials have very similar viscosity as a function of loading, which suggests that unlike the significant variations in particle size seen in water, the swelling by TEGDMA is more uniform regardless of the polysiloxane concentration. At low loading levels, the effect of the dispersed nanogel on TEGDMA viscosity is minimal. In the moderate loading range, both the particle concentration and the greater proportions of monomer that are taken up by infiltration into the nanogels lead to particle–particle interactions becoming dominant over resin–particle interactions. At 40 wt% nanogel loading, the viscosity is quite high since the matrix phase is approaching a continuous interphase rather than some combination of bulk TEGDMA and the interfacial regions around the monomer-swollen nanogels.
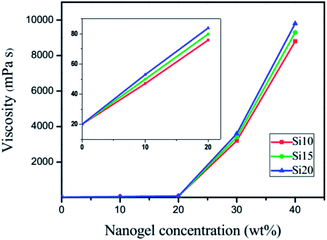 |
| | Fig. 3 Viscosity data at 15 °C for the nanogel added to TEGDMA at various loading levels from 0 to 40 wt%. | |
Kinetics of photopolymerization
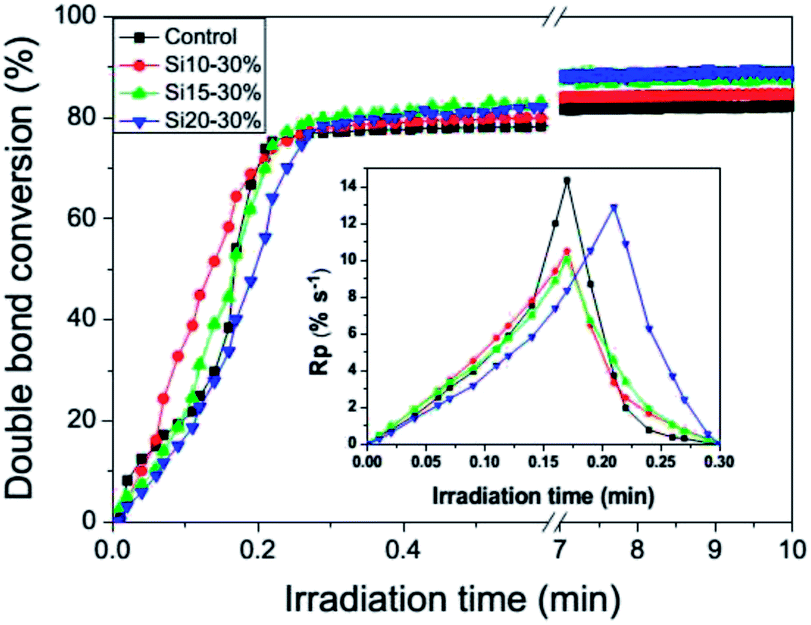
Photopolymerization kinetics plots with the accompanying rates are shown in Fig. 4. In general, the reaction profile of the TEGDMA control was similar to all the nanogel-modified resins, irrespective of the difference in polysiloxane content of the nanogel. The TEGDMA control shows typical auto-acceleration with a rate maximum that occurs relatively late in the conversion due to the significant mobility introduced by the triethylene glycol linkage. When the nanogels were loaded into TEGDMA, depending on the concentration used, two phases were presented: a bulk TEGDMA phase and a nanogel phase that is infiltrated by the monomer, which is facilitated by the solution polymerization process used to prepare the nanogel. This leads to a higher viscosity environment in the interior and interfacial regions of the nanogel phase. With the addition of the nanogels to TEGDMA as well as the increase in polysiloxane content in the nanogel, the reaction rate was modestly reduced while the limiting conversion was increased, which indicates greater late-stage mobility or delayed vitrification, probably induced by the polysiloxane segments within the nanogel domain.
 |
| | Fig. 4 Photopolymerization kinetics profiles and polymerization rate profiles for the control, Si10, Si15, and Si20 nanogel-modified materials at 30 wt% loading. All formulations were irradiated with 365 (±10) nm UV light at an intensity of 10 mW cm−2 with 0.5 wt% DMPA (relative to TEGDMA). | |
Shrinkage stress in photopolymerization
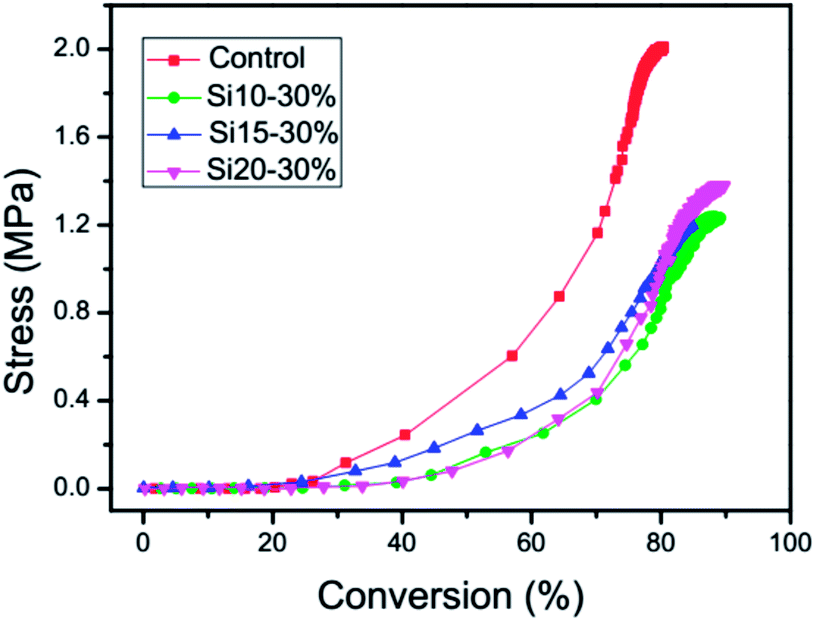
The baseline stress values of the ambient temperature UV-cured TEGDMA samples as controls were about 2 MPa. As shown in Fig. 5, the addition of nanogel to TEGDMA progressively decreases the final stress compared with the control. This is due primarily to the replacement of the monomer by the prepolymerized nanogel, which contributes to reduced overall reactive group concentrations and decreased volume changes during network formation. There is no clear trend in stress reduction potential based on the polysiloxane content of the nanogel additive. By correlating the polymerization stress with conversion through the simultaneous measurements (Fig. 6), it is clear that nanogel addition provides reduced final stress along with an increase in overall conversion. It is also evident that the resins with the three different nanogels all present a significant delay in the onset of stress compared with the control. Based on the kinetic results (Fig. 4), this behavior is not associated with preferential higher reactivity in the dispersed nanogel phase, as previously observed with other nanogel additives.25 Instead, due to a differential in modulus development between the nanogel and matrix phases as the polymerization proceeds, which would be expected, the bulk modulus development that contributes to stress may be concentrated in the later stages of the conversion.
 |
| | Fig. 5 Shrinkage stress for the TEGDMA monomer and the resins with variations in nanogel loading and the polysiloxane content within the nanogels. | |
 |
| | Fig. 6 Stress correlation with conversion for 30 wt% loading of various polysiloxane-modified nanogels in TEGDMA. The control is TEGDMA with 0.5 wt% DMPA. | |
Flexural strength and flexural modulus of photopolymerization materials
Results of the flexural strength and modulus measurements for the Si10, Si15 and Si20 systems with 0–40 wt% nanogel loading are shown in Fig. 7. The flexural strength, which is obtained in three-point bending mode, involves both tensile and compressive components in the stress field. The control system had a flexural strength of 78.6 MPa. When 10 wt% or 20 wt% of any of the nanogels was added into TEGDMA, the flexural strength was effectively maintained or only modestly reduced compared with the control. However, when 30 wt% or more of the nanogel was introduced into the resin, an obvious decrease in the flexural strength was observed, with the exception of the Si10 material at 30 wt% loading. The bulk flexural modulus, which is a critical component in the evolution of polymerization stress, was enhanced by nanogel incorporation up to 30 wt% loading. However, when 40 wt% of any of the nanogels was added, which is likely at or approaching a confluent nanogel configuration, the flexural modulus was decreased markedly. In general, there was less variability associated with the differences in polysiloxane content in the nanogel in terms of bulk modulus, but as with the flexural strength results there was no clear trend between the nanogels used.
 |
| | Fig. 7 Flexural strength (MPa) and modulus (GPa) tests for nanogel-modified TEGDMA resin and composite formulations. | |
Dynamic mechanical thermal analyses
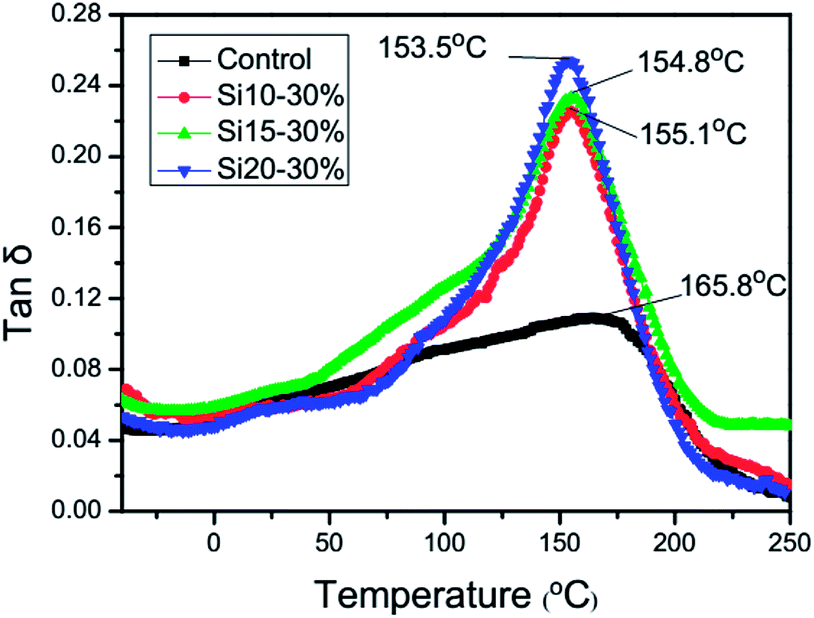
DMTA was employed to characterize the dynamic mechanical behavior of the fully cured TEGDMA homopolymer and the nanogel-modified materials. Compared with the control system, the addition of the nanogels reduces the polymeric Tg by approximately 10 °C with what appear to be slightly lower Tg values arising from the higher polysiloxane contents, which is opposite to the trend noted in the Tg values of the individual nanogels (Table 1). Under ambient curing conditions, it is evident that TEGDMA conversion in and around the monomer-swollen nanogel domains is higher than in the TEGDMA matrix. It is not known whether any conversion differential persists in the thermally post-cured samples, but these results do indicate that the ultimate Tg of the TEGDMA-infused nanogel is relatively close to that of the fully cured TEGDMA homopolymer. The nanogel construction involves relatively short but highly interconnected primary polymer chains, due to extensive chain transfer during nanoparticle formation. In spite of the high chain end concentration associated with the nanogel structures, the methacrylate groups present on the chain ends enable crosslinking within the nanogel and with the matrix to contribute to a relatively high Tg. When the nanogels were added to TEGDMA, the tanδ peak became narrower and its intensity increased, which indicated that the consolidation of the bulk TEGDMA and nanogel phases is effectively complete at the 30 wt% loading level tested here (Fig. 8).
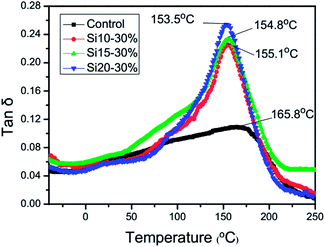 |
| | Fig. 8 tanδ behavior for the 30 wt% nanogel-modified systems using a 10 °C min−1 scan rate. | |
Nanogel self-floating ability
Fig. 9 shows the relative nitrogen concentrations as a means of following the nanogel concentration of the top and bottom layers with different standing times. The concentration of nitrogen in the top layers of Si10, Si15 and Si20 increased with the increase in standing time, and approached an equilibrium at about 90 min. This gives calculated mobility rates of about 19.1%, 23.4% and 30.5%, respectively, for the three different nanogels. The time-dependent nanogel concentrations in the bottom layer decreased in a coordinated manner as would be expected. It is clear that the self-floating ability of the nanogel was directly related to the increase in the polysiloxane content because of its effect on surface tension and density.
 |
| | Fig. 9 Relative concentration of nitrogen in the elemental analyses of the top and bottom layers of dichloromethane containing Si10, Si15 and Si20 at 10 wt% loading sampled at different standing times (assuming a relative concentration of 1 at 0 min). | |
Surface element analysis by XPS
Surface element analysis was employed to further investigate the self-floating ability of these nanogels when dispersed in the monomer. The abundance of C, O and Si on the surface of each segment sectioned from the polymer (PTEGDMA) rod with 30 wt% Si20 was detected by XPS, as shown in Fig. 10(a). The content of Si can represent the content of the nanogels because silicon is only present in the nanogels. From Fig. 10(b), it can be seen that the content of Si decreased gradually from the top to the bottom layer. For the Si20 nanogels at 30 wt% in the polymer, the content of Si as a function of increasing sample depth was 13.43%, 8.06%, 5.33% and 3.69%, which clearly documents a gradient distribution that can be attributed to the lower surface tension and density of the polysiloxane component of the dispersed nanogel particles. It is important to note that the gradient structured nanogel-modified monomers and polymers remain optically transparent, which suggests that these nanogels, with their hybrid organic–inorganic character, are not prone to significant aggregation either before or as a consequence of their polymerization within the forming matrix. Similar behaviour is revealed for the Si10 and Si15 nanogel-modified systems. The self-floating ability of Si10 is slightly weaker than those of Si15 and Si20 as a result of the lower content of polysiloxane in the nanogel.
 |
| | Fig. 10 (a) XPS survey spectrum of the PTEGDMA containing Si20 of 30 wt%; (b) silicon content in each layer of the PTEGDMA containing Si20, Si15 and Si10. | |
Surface morphology and elemental composition analysis by SEM and EDS
The surface morphologies of the polymer films containing 30 wt% Si10, Si15 and Si20, were observed by SEM. It is obvious that there are irregular protuberances on the surfaces of films with the nanogels that are not present on the surface of the PTEGDMA control. Further research on the irregular protuberances by EDS showed that the content of Si in these regions was higher than in other areas. It is therefore conjectured that the enrichment of the polysiloxane-modified nanogel resulted in the formation of the irregular protuberances. Furthermore, the number of irregular protuberances was generally increased and began to cluster as the content of polysiloxane in the nanogel increased. This further demonstrated that the increase in polysiloxane content in the nanogel contributes to improving the self-floating ability of the nanogel (Fig. 11).
 |
| | Fig. 11 SEM images of the polymer films (PTEGDMA) (a) without nanogel, (b) 30 wt% Si10 in TEGDMA, (c) 30 wt% Si15 in TEGDMA, (d) 30 wt% Si20 in TEGDMA, (the inset in (d) shows the content of Si in different areas by EDS). | |
The dispersion surface energy and contact angle of photopolymerization materials
The contact angle of a liquid water droplet on a surface is a direct reflection of the hydrophilicity/hydrophobicity of the surface. The dispersion surface energy (γdS) can be estimated by the geometric mean method on the basis of the contact angles. We calculated the dispersion surface energies of the polymer films from the Si10-, Si15- and Si20-containing materials based on the water contact angles observed on their respective surfaces. The equilibrium contact angle of a drop of liquid on a planar solid surface is determined based on three surface tensions. Here, γLV is the surface tension at the liquid–vapor interface, γSL is at the solid–liquid interface, and γSV is at the solid–vapor interface.| | |
γSL = γSV − γLVcosθ
| (1) |
According to Fowkes' study, the interface tension could also be calculated by the following formulation:
| | |
γSL = γS + γLV − 2(γdLγdS)1/2
| (2) |
Eqn (1) and (2) give:
| | |
γdS = [γLV(1 + cosθ)]2/4γdL
| (3) |
H
2O (
γLV = 72.7 mN m
−1,
γdL = 23.9 mN m
−1) was used as the testing liquid with its contact angle (
θ) on the planar surface of solid polymeric films evaluated here. From
θ, the
γdS values can be calculated
via eqn (3).
θ and values of
γdS are given in
Table 2.
Table 2 The contact angle and the dispersion surface energy (γdS) values of the gradient polymer films containing the nanogels
| Composite formula |
θ (H2O)/° |
γdS (H2O)/mN m−1 |
| TEGDMA |
43.5 |
164.5 |
| Si10 (30 wt%) |
60.7 |
122.7 |
| Si15 (30 wt%) |
70.6 |
98.2 |
| Si20 (30 wt%) |
84.4 |
66.7 |
The data in Table 2 show that at equivalent nanogel loading levels, higher concentrations of polysiloxane in the nanogel produced larger contact angles (θ) for H2O on the cured films along with smaller values of γdS. This is attributed to the enrichment of polysiloxane on the surface, leading to a lower surface energy, which is consistent with the SEM results.
Thermogravimetric analysis
Fig. 12 shows the peak temperatures at the maximum weight loss rates for the different layers of the polymers. The peak degradation temperature progressively increased from the bottom to the top layer, indicating a gradient in thermal stability that follows the gradient distribution of the polysiloxane-modified nanogel within the polymer. The polysiloxane offers the nanogel good thermal stability because of the relative strength of the Si–O bond compared with the C–C and C–O bonds. Along with the increase in thermostability associated with the nanogel loading level, an analogous stabilization effect was noted in terms of the polysiloxane content in the nanogel.
 |
| | Fig. 12 Peak temperatures at the maximum weight loss rates of each layer of PTEGDMA containing Si20, Si15 and Si10. | |
Conclusions
In this paper, three kinds of photopolymerizable polysiloxane-modified nanogels were synthesized and their structures were confirmed by 1H-NMR, FTIR, TEM and GPC. The nanogels were mixed with TEGDMA at various concentrations to evaluate a series of properties. The addition of polysiloxane-modified nanogels to the monomer at moderate loading levels was shown to reduce shrinkage stress without compromising the mechanical properties of the corresponding polymers. Most importantly, nanogels containing internal polysiloxane crosslink units had good self-floating abilities in the monomer, which allowed the spontaneous formation of well-controlled gradient structures before polymerization. The heterogeneous structure and the associated properties could be locked into the polymer by photopolymerization. The increase in polysiloxane content in the nanogel was found to improve its self-floating ability due to the lower surface tension and energy of polysiloxane. Polymers containing the nanogels presented gradients in thermal stability that depended on the nanogel loading and the polysiloxane concentration within the nanogel. The enrichment of the nanogels on the surfaces of the polymer films produced a significant decrease in the dispersion surface energy, along with the generation of more hydrophobic surfaces. Regulating the gradient properties of the polymer by using the polysiloxane-modified nanogels with self-floating ability is simple, convenient and environmentally friendly due to the combination of the advantages of photopolymerization technology, nanogels, and polysiloxane. The polysiloxane-modified nanogels have interesting potential for regulating the gradient properties and surface microstructure of polymers.
Acknowledgements
The financial support from the National Natural Science Foundation of China (Grant no. 51273014) is gratefully acknowledged along with additional support provided by NIH/NIDCR R01DE022348.
Notes and references
- L. V. Karabanova, S. V. Mikhalovsky and A. W. Lloyd, J. Mater. Chem., 2012, 22, 7919–7928 RSC.
- A. Ahmed, J. Smith and H. F. Zhang, Chem. Commun., 2011, 47, 11754–11756 RSC.
- R. R. Bhat, B. N. Chaney, J. Rowley, A. Liebmann-Vinson and J. Genzer, Adv. Mater., 2005, 17, 2802–2807 CrossRef CAS.
- B. Y. Wen, G. Wu and J. Yu, Polymer, 2004, 45, 3359–3365 CrossRef CAS PubMed.
- M. S. Kim, G. Khang and H. B. Lee, Prog. Polym. Sci., 2008, 33, 138–164 CrossRef CAS PubMed.
- J. Genzer and R. R. Bhat, Langmuir, 2008, 24, 2294–2317 CrossRef CAS PubMed.
- S. Morgenthaler, C. Zink and N. D. Spencer, Soft Matter, 2008, 4, 419–434 RSC.
- Y. Y. Cui, J. W. Yang, Z. H. Zeng, Z. Zeng and Y. L. Chen, Eur. Polym. J., 2007, 43, 3912–3922 CrossRef CAS PubMed.
- D. G. Yao, W. Zhang and J. G. Zhou, Biomacromolecules, 2009, 10, 1282–1286 CrossRef CAS PubMed.
- D. W. Jiang, X. Y. Huang, F. Qiu, C. P. Luo and L. L. Huang, Macromolecules, 2010, 43, 71–76 CrossRef CAS.
- C. Song, Z. Xu and J. Li, Mater. Des., 2007, 28, 1012–1015 CrossRef CAS PubMed.
- X. Peng, M. Yan and W. Shi, Scr. Mater., 2007, 56, 907–909 CrossRef CAS PubMed.
- N. B. Graham and A. Cameron, Pure Appl. Chem., 1998, 70, 1271–1275 CrossRef CAS.
- A. V. Kabanov and S. V. Vinogradov, Angew. Chem., Int. Ed., 2009, 48, 5418–5429 CrossRef CAS PubMed.
- N. B. Cramer, J. W. Stansbury and C. N. Bowman, J. Dent. Res., 2011, 90, 402–416 CrossRef CAS PubMed.
- T. T. Gan, Y. Guan and Y. J. Zhang, J. Mater. Chem., 2010, 20, 5937–5944 RSC.
- K. Okeyoshi, D. Suzuki and R. Yoshida, Langmuir, 2012, 28, 1539–1544 CrossRef CAS PubMed.
- C. D. Donahoe, T. L. Cohen, W. L. Li, P. K. Nguyen, J. D. Fortner, R. D. Mitra and D. L. Elbert, Langmuir, 2013, 29, 4128–4139 CrossRef CAS PubMed.
- R. R. Moraes, J. W. Garcia, M. D. Barros, S. H. Lewis, C. S. Pfeifer, J. C. Liu and J. W. Stansbury, Dent. Mater., 2011, 27, 509–519 CrossRef CAS PubMed.
- F. Sun, S. L. Jiang and J. Liu, Nucl. Instrum. Methods Phys. Res., Sect. B, 2007, 264, 318–322 CrossRef CAS PubMed.
- F. Sun, N. Zhang, J. Nie and H. G. Du, J. Mater. Chem., 2011, 21, 17290–17296 RSC.
- N. Zhang, M. L. Li, J. Nie and F. Sun, J. Mater. Chem., 2012, 22, 9166–9172 RSC.
- H. Lu, J. W. Stansbury, S. H. Dickens, F. C. Eichmiller and C. N. Bowman, J. Biomed. Mater. Res., Part B, 2004, 71, 206–213 CrossRef PubMed.
- J. C. Liu, I. Y. Rad, F. Sun and J. W. Stansbury, Polym. Chem., 2013, 5, 227–233 RSC.
- J. C. Liu, G. D. Howard, S. H. Lewis, M. D. Barros and J. W. Stansbury, Eur. Polym. J., 2012, 48, 1819–1828 CrossRef CAS PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c4ra02176b |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.