
Accelerated DNA recombination on a functionalized microfluidic chip
Abstract
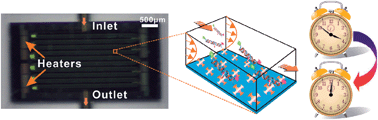
Genetic recombination is a powerful tool to create functional DNA hybrids with high controllability and precision for applications such as target gene therapy and molecular diagnostics, but it is often limited by the slow and low-efficient reconstruction of novel DNA species, especially in bulky volume solutions. Here, we present a microfluidic platform to enable rapid and consecutive on-line DNA digestion and ligation. The microdevice, embedded with a temperature sensor, offers controllable temperature and confined space to enhance intermolecular collision, and thus improves the slicing and splicing of DNA strands of interest. The stepwise functionalization of the microchannel surface with APTES, biotin and streptavidin permits the immobilization of biotinylated double stranded DNA (dsDNA) labeled with fluorescent probes at the ends for real-time monitoring. Digestion enzyme (BamH1) and DNA T4 ligase, subsequently, flow through the channel to hydrolyze and ligate the target DNA segments separately, and both digestion and ligation occur to a large extent within 10 min, with an efficiency of up to 71% (maximal at 1 h) and 63% (maximal at 2 h) of the highest value, respectively. Compared to conventional molecular diagnostics methods, which take several hours for digestion and ligation and deplete large volumes of reagents, such a system requires minimal time and sample volumes that are desirable in low-cost molecular therapy and point-of-care tests.
 Please wait while we load your content...
Please wait while we load your content...

