Ratiometric fluorescence probes based on a Michael acceptor type of coumarin and their application for the multichannel imaging of in vivo glutathione†
Gun-Joong Kima,
Doo-Ha Yoona,
Mi-Yeon Yunb,
Hyockman Kwonb,
Hyun-Joon Ha*a and
Hae-Jo Kim*a
aDepartment of Chemistry, Hankuk University of Foreign Studies, Yongin 449-791, Korea. E-mail: hjha@hufs.ac.kr; haejkim@hufs.ac.kr
bDepartment of Bioscience and Biotechnology, Hankuk University of Foreign Studies, Yongin 449-791, Korea
First published on 10th April 2014
Abstract
A series of Michael acceptors based on a coumarin moiety, were developed as fluorescent probes for ratiometric detection of in vivo glutathione. The α,β-unsaturated Michael acceptors were transformed into non-conjugated molecules through the Michael addition of biothiols. The resulting UV-vis and fluorescence spectra of the probes revealed characteristic ratiometric responses, which were successfully applied for the multichannel imaging of in vivo glutathione.
Introduction
Biothiols such as cysteine (Cys), homocysteine (Hcy), and glutathione (γ-glutamylcysteinylglycine, GSH) are involved in a myriad of vital cellular processes including redox homeostasis.1 However, abnormal levels of the biothiols are implicated in many human diseases, such as cancer and AIDS.2 Therefore, it is of great importance to detect the levels of biothiols both in vitro and in vivo. In spite of recent advances in the biothiol probes,3 however, there have been developed a few fluorophores that are able to detect the cellular biothiols free of the environmental and operational artifacts. One possible strategy for the artifact-free detection of biothiols is to introduce a self-calibrating module in the probe and to produce a ratiometric response.4Coumarin acts as a versatile scaffold for many fluorophores and was utilized as the Michael acceptors for detection of biothiols, but their ratiometric response in conjunction with reactivity as a Michael acceptor toward thiols has not been systematically studied yet. Herein we report a series of Michael acceptors (1–4) based on a coumarin moiety, which are expected to display ratiometric responses upon reaction with biothiols. The conjugated Michael acceptor type of probes are transformed into the non-conjugated molecules through the Michael addition of biothiols and induce characteristic ratiometric responses capable of the multichannel imaging of GSH in living cells.
Results and discussion
Coumarin aldehyde is a useful compound as the common and versatile reagent of aldol reaction. Whereas coumarin itself exhibits a strong fluorescence, coumarin derivatives possessing a carbonyl group and their vinyl analogues do not display a significant fluorescence. Their weak fluorescence may result from the existence of a quencher carbonyl group, which lowers the lowest unoccupied molecular orbital (LUMO) energy level of the fluorophore in order to absorb the excited electron and prohibits the excited electron from the radiative decay to the highest occupied molecular orbital (HOMO) level of the coumarin unit (Fig. S1†).5 For rational design of the biothiol probes, an aldol condensation reaction or Wittig reaction of coumarin aldehyde were simply carried out to afford a variety of electronically tunable conjugate enone fluorophores. A highly nucleophilic thiol group could react with the conjugated enone, which would lead to non-conjugated and fluorescent turn-on coumarin through the Michael addition reaction (Scheme 1). | ||
| Scheme 1 Plausible fluorescence turn-on mechanism of Michael acceptors upon reaction with a biothiol. | ||
The electronic properties of the probes (1–4) were tunable and readily available through aldol condensation or Wittig reaction of coumarin aldehyde from ester to diketo group (Scheme 2).
 | ||
| Scheme 2 Preparation of probes through an aldol condensation or Wittig reaction. | ||
The chemical reaction of the Michael acceptors with 2-mercaptoethanol (ME), an organic soluble model compound of biothiols, was observed by 1H NMR spectroscopy. Spectral analysis uncovered the position of the conjugate addition by a thiol group. Upon addition of ME, the spectra of 4 displayed another simple set of spectra within 5 min (Fig. 1). The vinylic proton (Hb) of 4 at 7.45 ppm disappeared with the concomitant appearance of a new peak at 4.49 ppm. On the other hand, relatively small chemical shifts of the aromatic protons indicated that the reaction took place in the peripheral region rather than in the aromatic regions of 4. The spectral analysis revealed the resulting simple set of spectra to be that of the expected Michael product. A similar spectral change was observed in the case of 2 (Fig. S7†).6 1H NMR experiments revealed that the thiol nucleophile, known as a relatively soft nucleophile, attacked the aliphatic vinylic position at the Michael acceptor site, although some hard nucleophiles such as cyanide would attack the aromatic region.7
 | ||
| Fig. 1 Partial 1H NMR spectra of 4 (20 mM) in DMSO-d6 upon addition of ME (2.5 equiv.). (A) 0 min, (B) 5 min. | ||
As expected, UV-vis spectra exhibited a profound ratiometric change when 2 was treated with GSH. Time-dependent UV-vis spectra of 2 (10 μM) were monitored in the presence of excess GSH (1.0 mM) in 50% DMSO–HEPES buffer (0.1 M HEPES, pH 7.4). While probe 2 showed a UV-vis absorption maximum centered at 458 nm (ε 4.2 × 104 M−1 cm−1), 2–GSH conjugate triggered a noticeable hypsochromic shift (ΔA −59 nm) with an isosbestic point at 416 nm. The rate constant of 2–GSH was calculated as kobs = 0.07 × 10−4 s−1 (τ 28 h) at 25 °C under the pseudo first-order reaction conditions.
On the other hand, the more activated Michael acceptor 4 was so rapidly reacted with GSH that the Michael addition reaction was complete within 5 min. Upon addition of 100 equiv. GSH (1.0 mM) to 4 (10 μM) in 50% DMSO–HEPES buffer (0.1 M HEPES, pH 7.4), a prominent hypsochromic shift was observed in the UV-vis spectra. While probe 4 showed a UV-vis absorption maximum centered at 464 nm (ε 4.8 × 104 M−1 cm−1), 4–GSH conjugate triggered a hypsochromic shift (ΔA −58 nm) to λmax 406 nm (ε 2.7 × 104 M−1 cm−1) with a clear isosbestic point at 423 nm (Fig. 2). The reaction kinetics analysis gave the calculated rate constant of 4-GSH with kobs 1.2 × 10−2 s−1 (τ 0.97 min) at 25 °C under the pseudo first-order reaction conditions.
 | ||
Fig. 2 Time-dependent UV-vis spectral changes upon addition of GSH (100 equiv.) to 10 μM of 4 in DMSO–HEPES buffer (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v, pH 7.4). Inset: their kinetics. :1, v/v, pH 7.4). Inset: their kinetics. | ||
We noticed that the Michael addition reaction was significantly affected by the substituents of α,β-unsaturated carbonyl compounds and therefore investigated the electronic effects of the Michael acceptors by changing R groups at the α-position of the Michael acceptors. Remarkably, an ester form (1) was not reactive with GSH under the given pseudo first-order reaction conditions. An aldehyde form (3) reacted with GSH faster than 2. The diketo form (4) was observed to be the most accelerated in the Michael reactions owing to the plausible double activation effect of the diketo groups on the conjugate addition reaction (Fig. 3).
 | ||
| Fig. 3 Reaction kinetics upon addition of GSH (100 equiv.) to 1–4 (10 μM) in DMSO–HEPES buffer (1:1, v/v, pH 7.4). Probe 1 (open square), 2 (open circle), 3 (filled square), 4 (filled circle). Inset: magnified kinetics of 4. | ||
The quantum yields were also measured using coumarin 6 as the reference compound8 (Table 1). The ester form (1) displayed the most significant value in the quantum yield relative to the other probes, but its reaction with GSH was too slow for the kinetics measurement. The quantum yields of probe–GSH conjugates were approximately evaluated 10 h after the incubation of probe with 100 equiv. of GSH. Though probes 2 and 3 exhibited relatively well enhanced quantum yields with 1.7 and 2.7-fold increases, respectively, a dramatic enhancement was observed with probe 4 + GSH with a 27-fold increase in the quantum yield, which was attributable to the role of the diketo group of 4 as a fluorescence quencher. The diketo group of 4 also played a critical role in the rate acceleration relative to the other probes. Probe 4 was very reactive to GSH with k2 12 M−1 s−1 at 25 °C. Probe 3 was relatively reactive to GSH but it was unstable and further oxidized in air. These data were summarized in Table 1. In terms of the quantum yields, reaction rate and stability, 2 and 4 were the most suitable candidates for in vivo GSH imaging.
| Entry | Probe | λabs (nm)/ε (104 M−1 cm−1) | λexb/λem (nm) | ΦFc | k2d (M−1 s−1) |
|---|---|---|---|---|---|
| a [Probe] = 10 μM and [GSH] = 5 mM in 50% DMSO–HEPES buffer (0.1 M, pH 7.4).b Excitation at an isosbestic point.c Quantum yield measured from coumarin 6 (ΦF 0.78) as a standard.d The second-order rate constant at 25 °C.e No reaction. | |||||
| 1 | 1 | 451/4.3 | 512 | 0.384 | — |
| 2 | 2 | 458/4.2 | 539 | 0.261 | — |
| 3 | 3 | 467/5.4 | 540 | 0.145 | — |
| 4 | 4 | 464/4.8 | 551 | 0.007 | — |
| 5 | 1 + GSH | — | — | — | NRe |
| 6 | 2 + GSH | 399/2.8 | 416/475 | 0.444 | 0.007 |
| 7 | 3 + GSH | 396/2.2 | 424/476 | 0.387 | 0.41 |
| 8 | 4 + GSH | 406/2.7 | 423/487 | 0.189 | 12 |
As observed in the UV-vis spectra, the fluorescence spectra of 4 (λex 423 nm) also exhibited a prominent blue shift (ΔF −64 nm) from λmax 551 nm to 487 nm upon addition of biothiols (GSH, Hcy and Cys) (Fig. 4A). The fluorescence intensity of 4 was changed from F 0.097 to 5.09 with a 52-fold increase in the presence of 5 mM GSH (Fig. 4B), whereas the other natural amino acids (AAs) with neutral, basic, or acidic side chains did not induce any significant fluorescence changes. The competitive experiments also showed that the fluorescence intensities of 4 + AA could be restored up to the value of 4 + GSH only by the addition of GSH to the mixtures of 4 and the other natural AAs.
 | ||
| Fig. 4 (A) Fluorescence spectral changes of 4 (10 μM) in HEPES (0.1 M, pH 7.4) upon addition of various amino acids (500 equiv.). (B) The competition graphs of GSH over various amino acids (AA, 500 equiv.). | ||
A sensitivity curve of 4 toward GSH was obtained by measuring the emission spectra of 4 (10 μM) in HEPES (0.1 M, pH 7.4) at λem 487 nm by varying the concentration of GSH. The fluorescence intensity of 4 increased linearly over the concentration ranges from 0.5 to 10 equiv. of GSH with the limit of detection (LOD) of 5.8 μM GSH at 3σ/m, where σ is a standard deviation of blank measurements without GSH and m is the value of slope from the linear plot of fluorescence intensity of 4 against GSH (Fig. 5).
 | ||
| Fig. 5 Fluorometric determination of limit of detection after the addition of GSH to 4 (10 μM, λex/λem 423/487 nm) in HEPES (0.1 M, pH 7.4). | ||
Probe 2 was applied for in vivo imaging of GSH,9 which is the most abundant cellular biothiol.10 For detection of GSH in cells, HeLa cells were treated with 5 μM of 2 for 0.5 h and washed 3 times with PBS. The images of the live cells were taken by using a confocal laser scanning microscope (CLSM). The resulting fluorescence images indicated that probe 2 was clearly expressed in cytoplasm (Fig. 6). Blue and green channel fluorescence images of 2-GSH were monitored by intrinsic cellular GSH. If the cells were pretreated with α-lipoic acid (LPA, 500 μM, 1 day), an enhancer of GSH,11 and then stained with 2 (5 μM, 0.5 h), the fluorescence intensities in the blue (λem 405–488 nm) and green (λem 488–559 nm) channels were strengthened, while the intensity in the red channel (λem 559–700 nm) remained almost constant. Upon treatment of the live cells with a scavenger of GSH, N-ethylmaleimide (NEM, 100 μM, 0.5 h),12 and then with 2 (5 μM, 0.5 h), strongly red fluorescence images were observable due to the enrichment of GSH-free 2, while the intensities in the blue/green channels were decreasing.
 | ||
| Fig. 6 Confocal laser scanning microscopic images of HeLa cells incubated with 2 (5.0 μM) upon treatment of LPA or NEM. | ||
In the case of probe 4, prominent ratiometric color changes were observed from green to blue by GSH but the color change in the red channel was not as clear as that of 2 (Fig. 7). These CLSM experiments clearly showed that 2 is a powerful fluorescence probe for the multichannel detection of in vivo GSH by ratiometric fluorescence.
 | ||
| Fig. 7 Confocal laser scanning microscopic images of HeLa cells incubated with 4 (5.0 μM) upon treatment of LPA or NEM. | ||
Conclusion
We designed a series of ratiometric fluorescence probes (1–4) based on a coumarin moiety for detection of in vivo GSH. Probes 2 and 4 exhibited rapid and ratiometric fluorescence responses to biothiols, owing to the Michael addition of a thiol group to the enone unit of the probes. The fast and large ratiometric responses of 2 and 4 afforded clear multichannel imaging for celluar GSH. We expect these Michael acceptor type of fluorescent probes to become useful as the ratiometric fluorophores capable of determining the concentration of biothiols in living cells.Experimental section
All fluorescence and UV-vis absorption spectra were recorded in FP 6500 fluorescence spectrometer and HP 8453 absorption spectrometer, respectively. 1H/13C NMR spectra were recorded at 400/100 MHz NMR spectroscopy. Mass spectra were recorded on G6401A MS-spectrometer. All experiments were carried out with commercially available reagents and solvents, and used without further purification, unless otherwise noted.UV-vis and fluorescence spectral measurement
A stock solution (10 mM) of 1, 2, 3, and 4 in DMSO was prepared and used by dilution with aqueous DMSO–HEPES or HEPES buffer (0.10 M, pH 7.4). For UV-vis experiment, a sample solution (10 μM) were prepared by mixing 2 μL of the stock solution of probe (10 mM in DMSO) with an appropriate amount of each amino acid and finally diluted with DMSO–HEPES or HEPES buffer to afford the desired concentration of probe and AA. Fluorescence spectra were also measured similarly with a slit width of 3 nm × 3 nm.Determination of quantum yields
Quantum yields were measured in 50% DMSO–HEPES buffer (0.10 M, pH 7.4) by the relative method using the following equation,where Φ, F, and A are quantum yield, fluorescence spectral area, and absorbance at the emission wavelength, respectively, while n is the refractive index of the solvent. The subscript “r” denotes the respective values of a reference compound, coumarin 6 (Φr 0.78 in ethanol).
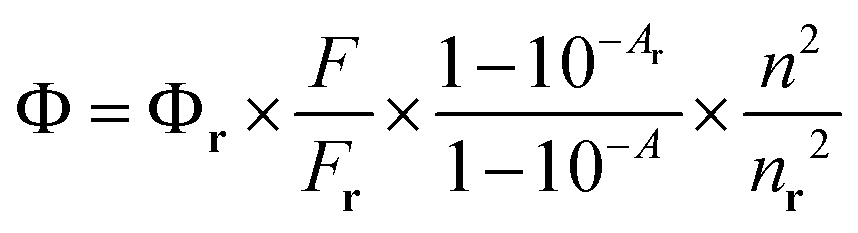
Fluorescence imaging of HeLa cells
For detection of biothiols in live cells, HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 100 units per mL penicillin, 100 mg mL−1 streptomycin, and 10% heat-inactivated fetal bovine serum. The cells were seeded on a Ø 35 mm glass-bottomed dish at the density of 0.8 × 105 cells in a culture medium and incubated overnight for live-cell imaging by confocal laser scanning microscopy (CLSM). The HeLa cells were treated with 5 μM 2 (or 4), which is prepared by dilution of 1.0 μL of stock solution of 2 (or 4) (10 mM in DMSO) with 2 mL of 1× PBS and incubated for 0.5 h and washed three times with pre-warmed 1× PBS before imaging by CLSM. In order to reduce the concentration of cellular GSH, HeLa cells were pretreated with NEM (100 μM, 0.5 h) prior to the incubation of 2 (or 4) (5 μM, 0.5 h). To increase the concentration of cellular GSH, the live cells were first incubated with LPA (500 μM, 24 h), followed by 2 (or 4) (5 μM, 0.5 h) and then washed 3 times with pre-warmed 1× PBS before imaging by CLSM at three channels (λex 380 nm, 488 nm, and 555 nm). After imaging the live cells, the mean fluorescence intensities at blue (λex 380 nm), green (λex 488 nm), and red (λex 555 nm) channels were measured in three different fields by ZEN imaging program. In order to reduce errors caused by background images outside of the cells, we also compared the intensity of the background image, but the level of the intensity was very low indicating that it did not seem to affect the mean fluorescence intensities.Synthesis of 1
7-(Diethylamino)-2-oxo-2H-chromene-3-carbaldehyde (0.096 g, 0.38 mmol)13 was dissolved in 3 mL of tetrahydrofuran. (Carbethoxymethylene)triphenylphosphorane (0.158 g, 0.454 mmol) was added to the above solution and then stirred at rt for 12 h. The mixture was evaporated under reduced pressure. Purification by column chromatography (EA–HEX = 1:2, v/v) provided the desired product as a yellow solid in 50% yield (0.060 g).
1H NMR (400 MHz, CDCl3): δ 7.69 (s, 1H) 7.52 (d, 1H, 3J = 16.0 Hz), 7.29 (d, 1H, 3J = 8.8 Hz), 6.99 (d, 1H, 3J = 16.0 Hz), 6.59 (d, 1H, 3J = 8.8 Hz, 4J = 2.4 Hz), 6.47 (d, 1H, 3J = 2.4 Hz), 4.23 (q, 2H, 3J = 7.2 Hz), 3.43 (q, 4H, 3J = 7.2 Hz), 1.31 (t, 3H, 3J = 7.2 Hz), 1.22 (t, 6H, 3J = 7.2 Hz). 13C NMR (100 MHz, CDCl3): δ 167.9, 160.4, 156.7, 151.9, 144.4, 139.5, 130.0, 119.7, 114.9, 109.6, 108.8, 97.2, 60.5, 45.2, 14.5, 12.7 (16 carbon peaks). HRMS (FAB+, m-NBA): m/z obsd 316.1545 ([M + H]+, calcd 316.1549 for C18H22NO4).
Synthesis of 2
7-(Diethylamino)-2-oxo-2H-chromene-3-carbaldehyde (0.15 g, 0.60 mmol) was dissolved in 1.8 mL of EtOH. Acetone (0.07 mL, 0.9 mmol) and piperidine (1 drop) were added at rt. The reaction mixture was refluxed for 6 h to afford a red solution, which was filtered and concentrated. Purification by column chromatography (DCM–EA–HEX = 1:2:7, v/v) furnished the desired product as an orange solid in 10% yield (0.017 g).
1H NMR (400 MHz, CDCl3): δ 7.77 (s, 1H) 7.46 (d, 1H, 3J = 16 Hz), 7.31 (d, 1H, 3J = 9.2 Hz), 7.13 (d, 1H, 3J = 16 Hz), 6.61 (dd, 1H, 4J = 2.4, 3J = 8.8 Hz), 6.48 (d, 1H, 4J = 2.4 Hz), 3.44 (q, 4H, 3J = 7.2 Hz), 2.35 (s, 3H), 1.23 (t, 6H, 3J = 7.2 Hz). 13C NMR (100 MHz, CDCl3): δ 198.7, 160.5, 156.6, 151.7, 144.1, 137.8, 129.9, 127.0, 114.3, 109.5, 108.7, 96.9, 45.0, 28.3, 12.4 (15 carbon peaks). HRMS (FAB+, m-NBA): m/z obsd 286.1440 ([M + H]+, calcd 286.1443 for C17H20NO3).
Synthesis of 3
7-(Diethylamino)-2-oxo-2H-chromene-3-carbaldehyde (0.2 g, 0.82 mmol) was dissolved in 10 mL of acetonitrile. (Triphenylphosphoranylidene)acetaldehyde (0.32 g, 1.05 mmol) and triethylamine (0.12 mL, 0.86 mmol) were added to the above solution and then were refluxed for 6 h. As the mixture was changed to red color, it was cooled to rt and then was evaporated under reduced pressure. Recrystallization of the crude product from ethanol afforded the desired product as a reddish solid in 80% yield (0.18 g).1H NMR (400 MHz, CDCl3): δ 9.64 (d, 1H, 3J = 7.7 Hz), 7.83 (s, 1H), 7.45 (d, 1H, 3J = 15.8 Hz), 7.34 (d, 1H, 3J = 8.8 Hz), 7.01 (dd, 1H, 3J = 7.7 Hz, 3J = 15.8 Hz), 6.63 (d, 1H, 3J = 8.8 Hz), 6.49 (s, 1H), 3.47 (q, 4H, 3J = 6.9 Hz), 1.25 (t, 6H, 3J = 6.9 Hz). 13C NMR (100 MHz, CDCl3): δ 194.1, 160.3, 157.1, 152.3, 147.3, 143.8, 130.4, 128.6, 113.9, 109.7, 108.6, 97.0, 45.1, 12.5 (14 carbon peaks). HRMS (FAB+, m-NBA): m/z obsd 272.1283 ([M + H]+, calcd 272.1287 for C16H18NO3).
Synthesis of 4
7-(Diethylamino)-2-oxo-2H-chromene-3-carbaldehyde (0.15 g, 0.60 mmol) was dissolved in 1.8 mL of EtOH. Acetylacetone (0.09 mL, 0.9 mmol) and piperidine (2 drops) were added and stirred at rt for 12 h to afford a red solution. The mixture was extracted with DCM. The combined organic layer was washed with H2O, dried over anhydrous MgSO4, filtered and concentrated under reduced pressure. Purification by column chromatography (DCM–EA–HEX = 1:2:7, v/v) furnished the desired product as an orange solid in 48% yield (0.96 g).
1H NMR (400 MHz, CDCl3): δ 7.78 (s, 1H) 7.59 (s, 1H), 7.28 (d, 1H, 3J = 9 Hz), 6.60 (dd, 1H, 4J = 2.6 Hz, 3J = 9 Hz), 6.46 (d, 1H, 4J = 2.6 Hz), 3.45 (q, 4H, 3J = 7 Hz), 2.44 (s, 3H), 2.36 (s, 3H) 1.23 (t, 6H, 3J = 7.2 Hz). 13C NMR (100 MHz, CDCl3): δ 205.5, 197.2, 161.1, 157.0, 152.1, 143.8, 141.7, 133.9, 130.6, 112.5, 109.6, 108.4, 96.9, 45.0, 31.4, 26.1, 12.4 (17 carbon peaks). HRMS (FAB+, m-NBA): m/z obsd 328.1544 ([M + H]+, calcd 328.1549 for C19H22NO4).
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) Grant funded by the Korean government (MICT) (NRF 2011-0028456) and the GRRC program of Gyeonggi province (GRRC HUFS-2013-B03).Notes and references
- (a) T. P. Dalton, H. G. Shertzer and A. Puga, Annu. Rev. Pharmacol. Toxicol., 1999, 39, 67 CrossRef CAS PubMed; (b) C. K. Mathews, K. E. van Holde and K. G. Ahern, Biochemistry, Addison-Wesley Publishing Co., San Francisco, 2000 Search PubMed.
- (a) D. M. Townsend, K. D. Tew and H. Tapiero, Biomed. Pharmacother., 2003, 57, 145 CrossRef CAS; (b) L. A. Herzenberg, S. C. De Rosa, J. G. Dubs, M. Roederer, M. T. Anderson, S. W. Ela, S. C. Deresinski and L. A. Herzenberg, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 1967 CrossRef CAS.
- (a) X. Yang, Y. Guo and R. M. Strongin, Angew. Chem., Int. Ed., 2011, 50, 10690 CrossRef CAS PubMed; (b) C. S. Lim, G. Masanta, H. J. Kim, J. H. Han, H. M. Kim and B. R. Cho, J. Am. Chem. Soc., 2011, 133, 11132 CrossRef CAS PubMed; (c) Z. Guo, S. W. Nam, S. Park and J. Yoon, Chem. Sci., 2012, 3, 2760 RSC; (d) M. H. Lee, J. H. Han, J.-H. Lee, H. G. Choi, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2012, 134, 17314 CrossRef CAS PubMed; (e) M. H. Lee, Z. Yang, C. W. Lim, Y. H. Lee, S. Dongbang, C. Kang and J. S. Kim, Chem. Rev., 2013, 113, 5071 CrossRef CAS PubMed; (f) Z. Guo, S. Park, J. Yoon and I. Shin, Chem. Soc. Rev., 2014, 43, 16 RSC.
- (a) B. Zhu, X. Zhang, Y. Li, P. Wang, H. Zhang and X. Zhuang, Chem. Commun., 2010, 46, 5710 RSC; (b) J. F. Zhang, C. S. Lim, S. Bhuniya, B. R. Cho and J. S. Kim, Org. Lett., 2011, 13, 1190 CrossRef CAS PubMed; (c) S.-Y. Lim and H.-J. Kim, Tetrahedron Lett., 2011, 52, 3189 CrossRef CAS PubMed; (d) G.-J. Kim, K. Lee, H. Kwon and H.-J. Kim, Org. Lett., 2011, 13, 2799 CrossRef CAS PubMed; (e) K. G. Reddie, W. H. Humphries, C. P. Payne, M. L. Kemp and M. Murthy, Org. Lett., 2012, 14, 680 CrossRef CAS PubMed; (f) J. Yao, K. Zhang, H. Zhu, F. Ma, M. Sun, H. Yu, J. Sun and S. Wang, Anal. Chem., 2013, 85, 6461 CrossRef CAS PubMed; (g) S.-Y. Lim, M.-J. Na and H.-J. Kim, Sens. Actuators, B, 2013, 185, 720 CrossRef CAS PubMed; (h) H. Lv, X.-F. Yang, Y. Zhong, Y. Guo, Z. Li and H. Li, Anal. Chem., 2014, 86, 1800 CrossRef CAS PubMed.
- (a) K.-S. Lee, T.-K. Kim, J. H. Lee, H.-J. Kim and J.-I. Hong, Chem. Commun., 2008, 6173 RSC; (b) G.-J. Kim and H.-J. Kim, Tetrahedron Lett., 2010, 51, 4670 CrossRef CAS PubMed; (c) S.-Y. Lim, S. Lee, S. B. Park and H.-J. Kim, Tetrahedron Lett., 2011, 52, 3902 CrossRef CAS PubMed; (d) H. Kwon, K. Lee and H.-J. Kim, Chem. Commun., 2011, 47, 1773 RSC.
- During our research, we found that a very similar structural probe (2–SPr conjugate) was reported as a ratiometric fluorophore for Hg(II) ions: W. Xuan, C. Chen, Y. Cao, W. He, W. Jiang, K. Liu and W. Wang, Chem. Commun., 2012, 48, 7292 RSC.
- (a) S. Park and H.-J. Kim, Sens. Actuators, B, 2012, 161, 317 CrossRef CAS PubMed; (b) G.-J. Kim and H.-J. Kim, Tetrahedron Lett., 2010, 51, 2914 CrossRef CAS PubMed.
- (a) J.-A. Richard, Y. Meyer, V. Jolivel, M. Massonneau, R. Dumeunier, D. Vaudry, H. Vaudry, P.-Y. Renard and A. Romieu, Bioconjugate Chem., 2008, 19, 1707 CrossRef CAS PubMed; (b) G. A. Reynolds and K. H. Drexhage, Opt. Commun., 1975, 13, 222 CrossRef CAS.
- (a) L. Yi, H. Li, L. Sun, L. Liu, C. Zhang and Z. Xi, Angew. Chem., Int. Ed., 2009, 48, 4034 CrossRef CAS PubMed; (b) N. Shao, J. Jin, H. Wang, J. Zheng, R. Yang, W. Chan and Z. Abliz, J. Am. Chem. Soc., 2010, 132, 725 CrossRef CAS PubMed; (c) J. H. Lee, C. S. Lim, Y. S. Tian, J. H. Han and B. R. Cho, J. Am. Chem. Soc., 2010, 132, 1216 CrossRef CAS PubMed; (d) L.-Y. Niu, Y.-S. Guan, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, J. Am. Chem. Soc., 2012, 134, 18928 CrossRef CAS PubMed.
- (a) A. Meister and M. E. Anderson, Annu. Rev. Biochem., 1983, 52, 711 CrossRef CAS PubMed; (b) M. E. Anderson, Chem.-Biol. Interact., 1998, 112, 1 CrossRef.
- (a) L. Packer, Drug Metab. Rev., 1998, 30, 245 CrossRef CAS PubMed; (b) L. Packer, H. J. Tritschler and L. Wessel, Free Radical Biol. Med., 1997, 22, 359 CrossRef CAS.
- C. R. Yellaturu, M. Bhanoori, I. Neeli and G. N. Rao, J. Biol. Chem., 2002, 277, 40148 CrossRef CAS PubMed.
- D.-N. Lee, G.-J. Kim and H.-J. Kim, Tetrahedron Lett., 2009, 50, 4766 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and mass spectra. See DOI: 10.1039/c4ra01933d |
| This journal is © The Royal Society of Chemistry 2014 |