 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDensity functional theory calculations on the CO catalytic oxidation on Al-embedded graphene
Q. G. Jiangab,
Z. M. Ao*c,
S. Lib and
Z. Wen*a
aKey Laboratory of Automobile Materials (Jilin University), Ministry of Education, School of Materials Science and Engineering, Jilin University, Changchun 130022, China. E-mail: wenzi@jlu.edu.cn
bSchool of Materials Science and Engineering, The University of New South Wales, Sydney, NSW 2052, Australia
cCentre for Clean Energy Technology, School of Chemistry and Forensic Science, University of Technology, Sydney, PO Box 123, Broadway, Sydney, NSW 2007, Australia. E-mail: zhimin.ao@uts.edu.au
First published on 7th April 2014
Abstract
The oxidation of CO molecules on Al-embedded graphene has been investigated by using the first principles calculations. Both Eley–Rideal (ER) and Langmuir–Hinshelwood (LH) oxidation mechanisms are considered. In the ER mechanism, an O2 molecule is first adsorbed and activated on Al-embedded graphene before a CO molecule approaches, the energy barrier for the primary step (CO + O2 → OOCO) is 0.79 eV. In the LH mechanism, O2 and CO molecules are firstly co-adsorbed on Al-embedded graphene, the energy barrier for the rate limiting step (CO + O2 → OOCO) is only 0.32 eV, much lower than that of ER mechanism, which indicates that LH mechanism is more favourable for CO oxidation on Al-embedded graphene. Hirshfeld charge analysis shows that the embedded Al atom would modify the charge distributions of co-adsorbed O2 and CO molecules. The charge transfer from O2 to CO molecule through the embedded Al atom plays an important role for the CO oxidation along the LH mechanism. Our result shows that the low cost Al-embedded graphene is an efficient catalyst for CO oxidation at room temperature.
1. Introduction
The oxidation of carbon monoxide (CO) has attracted great interest due to its importance in applications such as cleaning air and atmosphere purification for CO2 lasers,1,2 as well as removing CO from hydrogen gas fuel to avoid electrode poisoning in fuel cells.3 Some noble metals, such as Au,4 Pt,5–7 Pd6,8 and Rh9,10 have been studied as catalysts for CO oxidation, and the typical energy barriers for the rate limiting step in CO oxidation reaction reported are 0.46 eV for Au(221),4 0.79 eV (ref. 6) (0.82 eV (ref. 7)) for Pt(111), 0.91 eV (ref. 6) (0.93 eV (ref. 8)) for Pd(111), 1.17 eV (ref. 6) (1.01 eV (ref. 9)) for Rh(111), and 1.00 eV for Rh(100).10 Supported noble metal clusters are exploited to further decrease the reaction barriers for CO oxidation.11–15 However, these noble metal catalysts are costly and usually require high reaction temperature for efficient operation due to the high energy barriers, which is energy-consuming and may trigger explosion in the presence of hydrogen molecules. Therefore, developing low-cost catalysts for CO oxidation at room temperature is desirable, i.e. further reducing the energy barrier for the CO oxidation reaction using non-noble metals.As a novel form of carbon, graphene16 is a promising matrix to support metal atoms to realize new catalysts due to its outstanding electrical,17 mechanical18 and thermal properties.19 Moreover, the large surface-to-volume ratio also benefits for graphene as a support for heterogeneous catalysts. However, the interaction between pristine graphene and CO molecule is rather weak due to the inert nature of the graphene sheet.20 Theoretically, Au-, Fe-, Cu-, Pt- and Si-embedded graphenes were predicted to be highly active catalysts for CO oxidation,21–25 where the energy barriers for the rate limiting step are 0.31 eV for Au-embedded,21 0.58 eV for Fe-embedded,22 0.54 eV for Cu-embedded,23 0.59 eV for Pt-embedded,24 and 0.57 eV for Si-embedded graphene,25 respectively. Among these embedded graphenes, the Au-embedded graphene shows the best performance for CO oxidation. However, the adsorption energy of CO molecule (Ead_CO = −1.53 eV) is larger than that of O2 molecule (Ead_O2 = −1.34 eV) on Au-embedded graphene,21 which indicates that CO molecule has priority to adsorb on the Au-embedded graphene and may prevent the oxidation reaction. Therefore, further exploiting efficient graphene-based catalyst for CO oxidation is necessary. It has been reported that the reaction energy barrier is proportional to the adsorption energy of adsorbed molecules.6 For example, the reaction mechanism for CO oxidation on Au- and Cu-embedded graphene is the same, and the co-adsorption energy of CO and O2 molecules on Au-embedded graphene (Ead = −1.82 eV)21 is much smaller than that on Cu-embedded graphene (Ead = −3.29 eV),23 corresponding to a smaller energy barrier for the rate limiting step on Au-embedded graphene. Our previous studies show that Al-embedded graphene is a highly active catalyst for H2O dissociative adsorption26 and is a promising sensor material for CO detection.20 In addition, our calculated co-adsorption energy of CO and O2 molecules on Al-embedded graphene (Ead = −1.95 eV) is similar to that on Au-embeded graphene. Therefore, we expect that graphene embedded with the low-cost metal Al may be also highly active as a catalyst for the CO oxidation.
In this work, we will study the reaction mechanism of the CO oxidation on Al-embedded graphene through first principles calculations. The energy barriers for the reaction are discussed and the corresponding mechanisms are analyzed through understanding the electronic structures.
2. Calculation methods
The spin-unrestricted density functional theory (DFT) calculations are carried out by using Dmol3 package.27 Exchange−correlation functions are taken as generalized gradient approximation (GGA) with Perdew–Burke–Ernzerhof (PBE).28 DFT semicore pseudopotentials (DSPPs) core treatment is implemented for relativistic effects, which replaces core electrons by a single effective potential. Double numerical plus polarization (DNP) is employed as the basis set. The convergence tolerance of energy of 10−5 hartree is taken (1 hartree = 27.21 eV), and the maximal allowed force and displacement are 0.002 hartree per Å and 0.005 Å, respectively. It was reported that the selection of exchange−correlation functional has evidential effect on the result of adsorption energies. However, the effect on the calculated reaction energy barriers is much smaller.29 To investigate the minimum energy pathway for CO oxidation on graphene, linear synchronous transit/quadratic synchronous transit (LST/QST)30 and nudged elastic band (NEB)31 tools in Dmol3 module are used, which have been well validated to determine the structure of the transition state and the minimum energy reaction pathway. In the simulation, three-dimensional periodic boundary conditions are taken. The simulation cell consists of a 4 × 4 graphene supercell with a vacuum width of 20 Å above the graphene layer to minimize the interlayer interaction. The k-point is set to 5 × 5 × 1, and all atoms are allowed to relax according to previous reports.26 After structure relaxations, the density of states (DOS) are calculated with a finer k-point grid of 15 × 15 × 1. The DFT+D method within the Grimme scheme32 is used in all calculations to consider the van der Waals forces. The electron orbits of the free and adsorbed molecules are calculated with CASTEP code,33 where the ultrasoft pseudopotentials, GGA-PBE functional, an energy cutoff of 340 eV and 5 × 5 × 1 k-point meshes are used.For one molecule (CO or O2) adsorbed on graphene, the adsorption energy Ead is determined by,
| Ead = Emolecule/graphene − (Egraphene + Emolecule) | (1a) |
For two molecules (CO and O2) co-adsorbed on graphene, the co-adsorption energy Ead is determined by,
| Ead = Emolecules/graphene − (Egraphene + ECO + EO2) | (1b) |
3. Results and discussion
After embedding Al into graphene, where one C atom is substituted by one Al atom, the structure of graphene is reconstructed and the relaxed structure is shown in Fig. 1a. Covalent Al–C bonds are formed in graphene. Al is moved out of the plane to get more space due to its relatively large atomic radius of 1.25 Å, while that of C is 0.7 Å. The distance between the Al atom and graphene is hAl = 1.51 Å, which is consistent with the reported result of hAl = 1.43 Å.34 The C–Al bond length lC–Al is 1.85 Å, which is in agreement with the reported result of 1.85 Å (ref. 34 and 35) (1.86 Å (ref. 36)). The binding energy of the Al atom in graphene is −5.60 eV. The atomic charges obtained by Hirshfeld method near the dopant are also given in the configuration of IS in Fig. 1a, where the Al atom forms electron-deficiency position by transferring electrons (0.40e) to graphene. The strong interaction between the Al atom and C atoms can be further confirmed by the partial density of states (PDOS) (Fig. 1b), where significant overlap of the bands of the embedded Al atom and C atom is found. In addition, the Fermi level crosses the valence band, indicating the electron-deficiency character, which is consistent with the atomic charge distribution obtained by Hirshfeld method. | ||
| Fig. 1 (a) The pathway of the embedded Al atom diffusion on graphene, where IS, TS and FS represent initial structure, transition structure and final structure, respectively. The grey and pink atoms are C and Al in this and following figures. The atomic charge obtained by Hirshfeld analysis near the dopant is also given. (b) PDOS of Al atom and C atom on the Al-embedded graphene. The vertical lines indicate the Fermi level. | ||
To determine the possibility of Al aggregation, which is a problem when the concentration of the metals atoms is high,37 the diffusion of the Al atom to its neighbouring position on graphene is investigated (see Fig. 1a). It is found that the corresponding diffusion barrier for Al atom is 2.98 eV. Since a surface reaction at ambient temperature may occur only when the energy barrier is smaller than the critical barrier of Ecbar = 0.91 eV,38 the Al atom here is rather stable. In addition, the binding energy of Al on graphene is −5.60 eV, which is stronger than the cohesive energy of Al element of −3.39 eV per atom.39 Therefore, the Al dopant can disperse on graphene quite stably without clustering problem.
To investigate the oxidation of CO on Al-embedded graphene, the adsorptions of CO and O2 on Al-embedded graphene are first calculated, respectively. Fig. 2a shows the most stable configuration of a CO molecule adsorbed on Al-embedded graphene. It shows that the CO molecule is on the top of the embedded Al atom and vertical to graphene surface. Chemical bond is formed between the C atom in CO and the Al atom, indicating chemical adsorption of the CO molecule on Al-embedded graphene, which also agrees with the reported result.20 The corresponding adsorption energies and structure data of the most stable configurations for CO and O2 adsorption on Al-embedded graphene are listed in Table 1. The adsorption energy of a CO molecule on Al-embedded graphene Ead = −0.83 eV, and ∼0.15e is transferred from the CO molecule to the Al-embedded graphene based on Hirshfeld analysis. Fig. 2b shows the PDOS and orbitals of a free CO molecule. All orbitals are labelled and the 2π* anti-bond orbital is fully empty since it locates far above Fermi level, which is consistent with the literature results for CO molecule.5 The other three orbitals are fully occupied. In addition, the 5σ orbital with two electrons at the Fermi level state mainly locates on the C atom, indicating that CO always acts as a donor when its C atom pointing to the graphene surface.40 This agrees with the above Hirshfeld analysis result. Fig. 2c and d show PDOS and orbitals of the adsorbed CO molecule on Al-embedded graphene, respectively. Compared with the free CO molecule, the 5σ spin down peak is significantly depressed in the CO adsorbed on Al-embedded graphene due to the transferring of electrons from CO to Al atom. Because CO-2π* is close to the Fermi level, there is some electrons transferred from Al atom to CO-2π* state and CO-2π* state is partial filled as shown in Fig. 2c and d, which leads to the slight elongation of the C–O bond from 1.14 Å in free CO molecule to 1.15 Å.
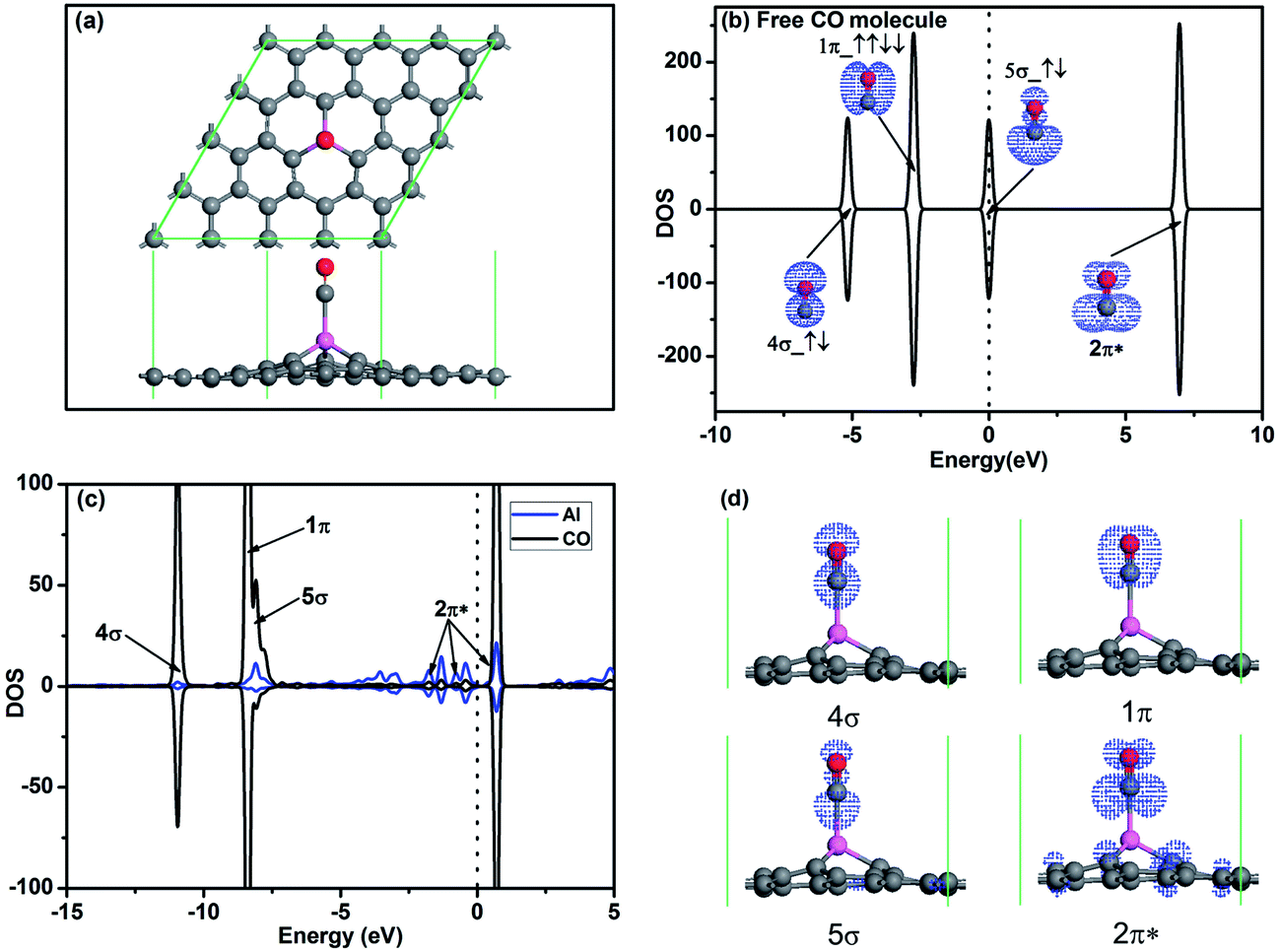 | ||
| Fig. 2 (a) The most stable structure of CO adsorbed on Al-embedded graphene. (b) PDOS and orbitals of the CO molecule, where the number of electrons for each orbital is also shown by arrows. (c) PDOS of CO molecule and the Al atom for the Al-embedded graphene with adsorbed CO. (d) Charge density of 4σ, 1π, 5σ and 2π* orbitals of CO molecule on Al-embedded graphene. The vertical lines indicate the Fermi level. | ||
| Ead (eV) | h (Å) | l (Å) | Q (e) | |
|---|---|---|---|---|
| Free CO | — | — | 1.14 | — |
| Adsorbed CO | −0.83 | 2.06 | 1.15 | 0.15 |
| Free O2 | — | — | 1.23 | — |
| Adsorbed O2 | −1.57 | 1.86 | 1.43 | −0.31 |
For the O2 adsorption on Al-embedded graphene, the O2 molecule is adsorbed on Al atom much stronger with an Ead of −1.57 eV and the O–O bond is slightly titled with the graphene sheet (see IS in Fig. 3a and d). To assess the stability of O2 molecule on graphene, we also studied the dissociative adsorption of an O2 molecule on Al-embedded graphene as shown in Fig. 3a. After dissociative adsorption, one O atom adsorbs on Al atom while the other O atom co-binds with the Al atom and a neighbouring C atom at FS state as shown in Fig. 3a. After LST/QST and NEB calculations, the energy barrier for the dissociative adsorption of an O2 molecule on the Al-embedded graphene is 1.66 eV > Ecbar = 0.91 eV,38 which indicates that the adsorbed O2 molecule is stable on Al-embedded graphene without dissociation at room temperature. The possibility of CO dissociation on Al-embedded graphene is also considered. Our DFT calculations show that binding energy of C–O bond (Eb = 11.57 eV) is much stronger than that of O–O bond (Eb = 6.36 eV). Thus CO should be more difficult to be dissociated. As seen in Fig. 3b, all the orbitals of free O2 molecule are labelled while the 2π* anti-bond orbital is half filled, which is consistent with the literature results for O2 molecule.41 When one O2 molecule is adsorbed on Al-embedded graphene, based on Hirshfeld analysis, there are about 0.31e to be transferred from the Al-embedded graphene to O2 molecule, which occupies the initial empty component of the O2–2π* orbital. This can be confirmed by two new peaks presence for O2–2π* orbital after its adsorption on the Al-embedded graphene (see Fig. 3c), which also leads to the elongation of the O–O bond from 1.23 Å in free O2 molecule to 1.43 Å. The hybridization between the Al atom and O2–2π* orbitals is observed near Fermi level from the PDOS (see Fig. 3c). The above results reveal that Al-embedded graphene has strong interactions with O2 and CO (corresponding Ead are −1.57 and −0.83 eV, respectively), but the adsorption of O2 is much stronger. The adsorbed O2 is efficiently activated, which facilitates the further oxidation of CO molecules.
 | ||
| Fig. 3 (a) The reaction pathway for the dissociative adsorption of O2 molecule on Al-embedded graphene. (b) PDOS and orbitals of the O2 molecule, where the number of electrons for each orbital is also shown by arrows. (c) PDOS of O2 molecule and Al atom for the Al-embedded graphene with adsorbed O2. (d) Charge density of 4σ, 5σ, 1π and 2π* orbitals of O2 molecule on Al-embedded graphene. The vertical lines indicate the Fermi level. | ||
Two mechanisms for CO oxidation have been established: Eley–Rideal (ER) mechanism and Langmuir–Hinshelwood (LH) mechanism.21–25,42,43 In the ER mechanism, the pre-adsorbed O2 molecule is activated by charge transferring with the substrate and an intermediate product forms when a free CO molecule approaching. In the LH mechanism, O2 and CO molecules co-adsorb on the embedded graphene before the reaction happening. Since O2 has stronger adsorption energy (−1.57 eV) than that of CO on Al-embedded graphene (−0.83 eV), the adsorption of O2 on Al-embedded graphene has higher priority, and thus the ER mechanism seems to be more favourable. Although the adsorption energy difference between CO and O2 is relatively large, the lower co-adsorption energy of CO and O2 molecules (−1.95 eV) indicates that there is a certain probability of having O2 and CO co-adsorbed on Al atom as discussed in literatures.21,23,24 Therefore, both mechanisms will be investigated in this work.
In order to determine the preferred mechanism for CO oxidation, the first reaction step of both ER and LH mechanisms is studied, respectively. If the ER mechanism is favourable, the physisorbed CO molecule will react with the pre-adsorbed O2 molecule to form OOCO intermediate (shown as the TS in Fig. 4a). If the LH mechanism is favourable, the physisorbed CO molecule will co-adsorb on Al atom with O2 molecule with a small energy barrier. The energy profile for the ER mechanism is shown in Fig. 4a. The configuration of physisorbed CO perpendicular to the graphene surface was selected as the initial state IS after considering all possible adsorption positions. When one CO molecule approaches the activated O2, after overcoming an energy barrier of 0.79 eV, one O–Al bond is broken, the O with dangling bond would form a covalent bond with the C atom in the CO molecule (see TS in Fig. 4a). Then the C atom of the CO binds with one C atom closed to the Al dopant and a peroxo-type OOCO complex is formed over the Al atom (see FS in Fig. 4a). This process is exothermic and releases energy of 1.28 eV. Fig. 4b shows the energy profile for the CO molecule co-adsorbed on Al atom with O2 molecule, where the CO molecule is titled towards the Al atom at TS state. After overcoming a small energy barrier of 0.03 eV, the CO molecule co-adsorbs on Al atom at FS state as shown in Fig. 4b. This process is exothermic with releasing energy of 0.18 eV.
 | ||
| Fig. 4 (a) The primary reaction step for the oxidation of CO molecule on Al-embedded graphene for ER mechanism. (b) The reaction pathway for the adsorption of CO molecule on Al-embedded graphene with pre-adsorbed O2 molecule. | ||
The energy barriers above are considered at zero Kelvin. To take into consideration the effect of temperature, free energy change (ΔG) between IS and TS is considered as the corrected energy barrier E′bar and can be determined by ΔG = ΔH − TΔS, where ΔH is the change in enthalpy, T is the room temperature (298.15 K), and ΔS is the change in entropy. It is known that ΔH = (ΔU + PΔV), ΔU = (ΔEtot + ΔEvib + ΔEtrans + ΔErot) and ΔS = ΔSvib + ΔStrans + ΔSrot, where ΔU is the change of internal energy, ΔEtot is the total electronic energy change obtained from DFT calculations, the subscript vib, trans and rot indicate the components from vibration, translation and rotation, respectively. All the above terms can be calculated with vibrational frequency calculations in Dmol3 code. The corrected energy barriers at room temperature for each step in Fig. 4 are: E′bar = 0.95 eV in Fig. 4a, and E′bar = 0.16 eV in Fig. 4b. This result is reasonable since the physisorbed CO molecule prefers to desorb from graphene at higher temperature, which makes the reaction more difficult to occur. Since a surface reaction at room temperature may occur only when Ebar < Ecbar = 0.91 eV,38 the reaction for the co-adsorption of CO and O2 molecules in Fig. 4b will happen at room temperature, the LH mechanism is thus focused in the following.
For CO oxidation with LH mechanism, there are several steps and also intermediate produces (MS) for the oxidation procedure as reported.21,23,24 For each reaction step, e.g. from IS to MS in Fig. 5a, there is also a transition state, the corresponding atomic structures before (IS) and after oxidation (FS), and MS are given in Fig. 5a. The configuration of co-adsorbed CO and O2 on the Al-embedded graphene is taken as the initial state (IS in Fig. 5a) before oxidation based on the discussion above, where CO and O2 are titled and parallel to the graphene surface, respectively. At TS state, one Al–O bond elongates from 1.86 to 2.38 Å and the new C–O bond is formed. After overcoming an energy barrier of 0.32 eV, OOCO complex (MS in Fig. 5a) is formed while one O–Al bond is broken. This reaction continues with the formation of one CO2 molecule (MS2) after overcoming an energy barrier of 0.18 eV (see Fig. 5b), and the CO2 molecule is physically adsorbed in MS2 structure with an adsorption energy of −0.16 eV. Since the energy released in this step (2.34 eV) can sufficiently surmount the adsorption energy, the desorption of the first produced CO2 molecule would be efficient. Note that the OOCO configuration in MS remains at TS2 with slight adjustments of bond length and bond angle. The remaining O atom will react with another CO to produce CO2 after overcoming an energy barrier of 0.10 eV based on our calculation (see Fig. 5c), similar to the reported result on Au-embedded graphene.21 The energy released in this step (2.60 eV) can also overcome the adsorption energy of CO2 (−0.40 eV) in FS structure and the desorption of the second CO2 molecule would be efficient as well. The energy profile for the formation of the first CO2 molecule is shown in Fig. 5d where the rate limiting energy barrier is quite low (0.32 eV) for the formation of OOCO complex. Therefore, CO molecules can be easily oxidized on Al-embedded graphene at room temperature, indicating that Al-embedded graphene is an excellent catalyst for the CO oxidation. The catalyst efficiency is the second highest among present catalysts except Au-embedded graphene (0.31 eV (ref. 21)).
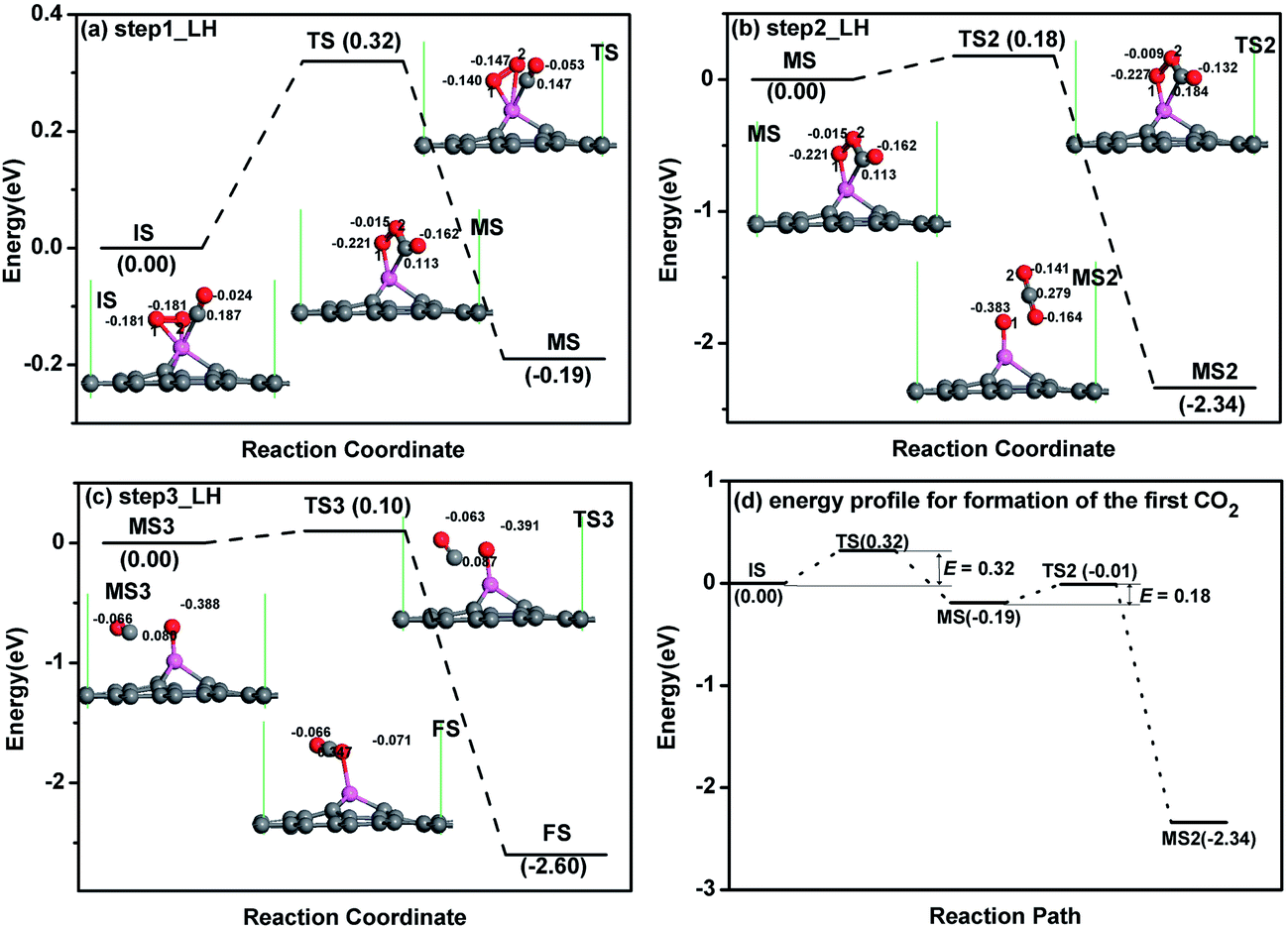 | ||
| Fig. 5 The reaction pathway of CO oxidation on Al-embedded graphene for LH mechanism: step1 (a), step2 (b) and step3 (c). The energy profile for the formation of the first CO2 molecule is shown in (d). The unit of E is eV, where E is the energy barrier. The Hirshfeld charge near the adsorbate is also shown. | ||
The corrected energy barriers at room temperature for each step in Fig. 5 are: E′bar = 0.35 eV for step1, E′bar = 0.12 eV for step2, and E′bar = 0.19 eV for step3. The energy barriers for step1 and step3 slightly increase at room temperature compared with those at zero kelvin. The reaction time for each step in Fig. 5 at room temperature is estimated by the following equation,
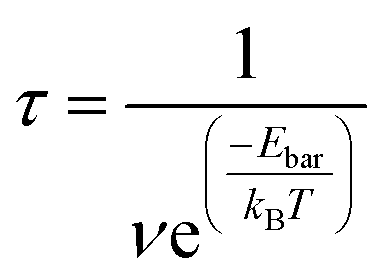 | (2) |
To gain more insight into the origin of the high activity of the Al-embedded graphene, the Hirshfeld charge transfer and PDOS near the adsorbates along the LH reaction path are shown in Fig. 5 and 6, respectively. O2 obtains 0.362e while CO loses 0.163e by Hirshfeld charge analysis for IS structure in Fig. 5a. The initial empty component of the O2-2π* orbital is partially filled while the CO-2π* orbital is nearly empty (see Fig. 6a) due to the electron transformation during the adsorption. Therefore, there is a tendency of charge transfer from the electron-efficiency O2 to the electron-inefficiency CO, which will facilitate the bond formation of O2 and CO. At TS state, the Hirshfeld charge of O2 molecule is 0.287e while that of CO molecule is 0.094e in Fig. 5a, which indicates that there is charge transfer from O2 to CO. The CO–2π* orbital is more filled and splitted as shown in Fig. 6b. After overcoming the energy barrier, there is more charge transfer from O2 to CO, where the Hirshfeld charge of O2 molecule is 0.237e while that of CO molecule is 0.049e for MS structure in Fig. 5a. The O2–2π* orbital is significantly depressed while the CO–2π* orbital is half filled in Fig. 6c. The above discussions show that the Al-embedded graphene mediate the charge distribution of O2 and CO molecules, where the charge transfer from O2 to CO plays a key role for the OOCO formation. Moreover, the states of C–O in CO molecule and O1–O2 (labelled in Fig. 5a) in O2 molecule interact with each other, strengthening C–O2 bond while weakening O1–O2 bond, as indicated by the superposition of the C–O and O1–O2 states at TS2 (see Fig. 6d), which will facilitate the formation of CO2 molecule.
 | ||
| Fig. 6 PDOS of adsorbed CO, O2 and Al atom for the IS (a), TS (b), MS (c) and TS2 (d). Black, red and pink curves represent PDOS of adsorbed CO, O2 and Al atom, respectively. The orbitals of the adsorbed CO and O2 molecules are roughly labelled. The vertical lines indicate the Fermi level. | ||
4. Conclusion
The oxidation of CO molecule on Al-embedded graphene has been investigated by using the first principles method. It is found that the LH mechanism is preferred for CO oxidation on Al-embedded graphene, where the energy barrier for the rate limiting step is only 0.32 eV. This barrier is lower than almost all reported catalysts, except the Au-embedded graphene with barrier of 0.31 eV. The charge transfer from O2 to CO molecule through the embedded Al plays an important role for the CO oxidation along the LH mechanism. Our result indicates that Al-embedded graphene can be a low cost and efficient catalyst for CO oxidation.Acknowledgements
We acknowledge supports by National Key Basic Research, Development Program (Grant no. 2010CB631001) and China Scholarship Council (no. 201206170088). ZA acknowledges the financial supports from the Chancellor's Postdoctoral Research Fellowship Program of the University of Technology, Sydney, and the Goldstar Award of the University of New South Wales (RG124422). SL would also like to thank the financial support from Australia Research Council (FT FT100100956).References
- A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Science, 2008, 321, 1331 CrossRef CAS PubMed.
- X. W. Xie, Y. Li, Z. Q. Liu, M. Haruta and W. J. Shen, Nature, 2009, 458, 746 CrossRef CAS PubMed.
- A. Hornes, A. B. Hungria, P. Bera, A. L. Camara, M. Fernandez-Garcia, A. Martinez-Arias, L. Barrio, M. Estrella, G. Zhou, J. J. Fonseca, J. C. Hanson and J. A. Rodriguez, J. Am. Chem. Soc., 2010, 132, 34 CrossRef CAS PubMed.
- Z. P. Liu, P. Hu and A. Alavi, J. Am. Chem. Soc., 2002, 124, 14770 CrossRef CAS PubMed.
- K. Bleakley and P. Hu, J. Am. Chem. Soc., 1999, 121, 7644 CrossRef CAS.
- X. Q. Gong, Z. P. Liu, R. Raval and P. Hu, J. Am. Chem. Soc., 2004, 126, 8 CrossRef CAS PubMed.
- C. Dupont, Y. Jugnet and D. Loffreda, J. Am. Chem. Soc., 2006, 128, 9129 CrossRef CAS PubMed.
- C. J. Zhang and P. Hu, J. Am. Chem. Soc., 2001, 123, 1166 CrossRef CAS PubMed.
- A. H. Zhang, J. Zhu and W. H. Duan, J. Chem. Phys., 2006, 124, 234703 CrossRef CAS PubMed.
- D. J. Liu, J. Phys. Chem. C, 2007, 111, 14698 CAS.
- Z. P. Liu, X. Q. Gong, J. Kohanoff, C. Sanchez and P. Hu, Phys. Rev. Lett., 2003, 91, 266102 CrossRef.
- B. Huber, P. Koskinen, H. Häkkinen and M. Moseler, Nat. Mater., 2006, 5, 44 CrossRef CAS PubMed.
- D. Y. Tang and J. Zhang, RSC Adv., 2013, 3, 15225 RSC.
- P. Ghosh, M. F. Camellone and S. Fabris, J. Phys. Chem. Lett., 2013, 4, 2256 CrossRef CAS.
- Y. G. Wang, Y. Yoon, V. A. Glezakou, J. Li and R. Rousseau, J. Am. Chem. Soc., 2013, 135, 10673 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Nature, 2005, 438, 197 CrossRef CAS PubMed.
- C. G. Lee, X. D. Wei, J. W. Kysar and J. Hone, Nature, 2008, 321, 385 CAS.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902 CrossRef CAS PubMed.
- Z. M. Ao, J. Yang, S. Li and Q. Jiang, Chem. Phys. Lett., 2008, 461, 276 CrossRef CAS PubMed.
- Y. H. Lu, M. Zhou, C. Zhang and Y. P. Feng, J. Phys. Chem. C, 2009, 113, 20156 CAS.
- E. H. Song, Z. Wen and Q. Jiang, J. Phys. Chem. C, 2011, 115, 3678 CAS.
- Y. F. Li, Z. Zhou, G. T. Yu, W. Chen and Z. F. Chen, J. Phys. Chem. C, 2010, 114, 6250 CAS.
- Y. N. Tang, Z. X. Yang and X. Q. Dai, Phys. Chem. Chem. Phys., 2012, 14, 16566 RSC.
- J. X. Zhao, Y. Chen and H. G. Fu, Theor. Chem. Acc., 2012, 131, 1 CrossRef CAS PubMed.
- Q. G. Jiang, Z. M. Ao and Q. Jiang, Phys. Chem. Chem. Phys., 2013, 15, 10859 RSC.
- B. Delley, J. Chem. Phys., 2000, 113, 7756 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- A. Roldán, J. M. Ricart and F. Illas, Theor. Chem. Acc., 2009, 123, 119 CrossRef.
- T. A. Halgren and W. N. Lipscomb, Chem. Phys. Lett., 1977, 49, 225 CrossRef CAS.
- G. Henkelman and H. Jonsson, J. Chem. Phys., 2000, 113, 9978 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed.
- M. D. Segall, P. L. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717 CrossRef CAS.
- J. Y. Dai, J. M. Yuan and P. Giannozzi, Appl. Phys. Lett., 2009, 95, 232105 CrossRef PubMed.
- J. Y. Dai and J. M. Yuan, Phys. Rev. B: Condens. Matter Mater. Phys, 2010, 81, 165414 CrossRef.
- P. A. Denis, Chem. Phys. Lett., 2010, 492, 251 CrossRef CAS PubMed.
- Z. M. Ao and F. M. Peeters, Phys. Rev. B: Condens. Matter Mater. Phys, 2010, 81, 205406 CrossRef.
- D. C. Young, Computational Chemistry: A Practical Guide for Applying Techniques to Real World Problems, Wiley, New York, 2001 Search PubMed.
- http://www.knowledgedoor.com/2/elements_handbook/cohesive_energy.html.
- O. Leenaerts, B. Partoens and F. M. Peeters, Phys. Rev. B: Condens. Matter Mater. Phys, 2008, 77, 125416 CrossRef.
- K. Honkala and K. Laasonen, Phys. Rev. Lett., 2000, 84, 705 CrossRef CAS.
- L. M. Molina and B. Hammer, J. Catal., 2005, 233, 399 CrossRef CAS PubMed.
- W. An, Y. Pei and X. C. Zeng, Nano Lett., 2008, 8, 195 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |